
Indoor Bacterial and Fungal Burden in “Moldy” versus “Non-Moldy” Homes: A Case Study Employing Advanced Sequencing Techniques in a US Metropolitan Area


 and
and 
Abstract
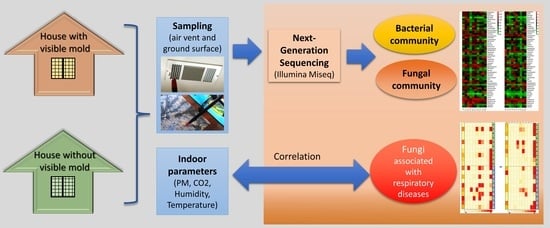
1. Introduction
2. Materials and Methods
2.1. Study Area and Sampling Plan
2.2. Measurement of Indoor Environment Parameters
2.3. DNA Extraction, Purification, and Sequencing
2.4. Sequence Processing and Analysis
2.5. PCR Detection of Fungal Species Known to Be Associated with Respiratory Complications
2.6. Statistical Analysis
3. Results
3.1. Data Showed the Presence of Several Pathogenic Bacterial and Fungal Genera with Distinctive Patterns in the Ground versus Air Vent Dust
3.2. Network Analysis Shows Higher Variability in Fungal Than Bacterial Communities in Dust Samples from Air Vents and Ground
3.3. The Fungal Communities in Air Vent Samples Differ Significantly between Home Types
3.4. Respiratory Disease-Related Fungal Species Were Detected in Both Home Types, Regardless of the Visibility of Fungi (Molds) in the Home
3.5. Respiratory Disease-Associated Fungi Did Not Show Any Significant Correlation with Indoor Environmental Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelley, S.T.; Gilbert, J.A. Studying the microbiology of the indoor environment. Genome Biol. 2013, 14, 202. [Google Scholar] [CrossRef]
- Horve, P.F.; Lloyd, S.; Mhuireach, G.A.; Dietz, L.; Fretz, M.; MacCrone, G.; Van Den Wymelenberg, K.; Ishaq, S.L. Building upon current knowledge and techniques of indoor microbiology to construct the next era of theory into microorganisms, health, and the built environment. J. Expo. Sci. Environ. Epidemiol. 2020, 30, 219–235. [Google Scholar] [CrossRef]
- Leung, M.H.; Tong, X.; Lee, P.K. Indoor microbiome and airborne pathogens. Compr. Biotechnol. 2019, 6, 96–106. [Google Scholar]
- Cox, J.; Stone, T.; Ryan, P.; Burkle, J.; Jandarov, R.; Mendell, M.J.; Adams, R.I.; Niemeier-Walsh, C.; Reponen, T. Associations of observed home dampness and mold with the fungal and bacterial dust microbiomes. Environ. Sci. Process. Impacts 2021, 23, 491–500. [Google Scholar] [CrossRef]
- Pausan, M.-R.; Blohs, M.; Mahnert, A.; Moissl-Eichinger, C. The sanitary indoor environment—A potential source for intact human-associated anaerobes. npj Biofilms Microbiomes 2022, 8, 44. [Google Scholar] [CrossRef]
- Emerson, J.B.; Keady, P.B.; Brewer, T.E.; Clements, N.; Morgan, E.E.; Awerbuch, J.; Miller, S.L.; Fierer, N. Impacts of flood damage on airborne bacteria and fungi in homes after the 2013 colorado front range flood. Environ. Sci. Technol. 2015, 49, 2675–2684. [Google Scholar] [CrossRef]
- Karvonen, A.M.; Hyvärinen, A.; Korppi, M.; Haverinen-Shaughnessy, U.; Renz, H.; Pfefferle, P.I.; Remes, S.; Genuneit, J.; Pekkanen, J. Moisture damage and asthma: A birth cohort study. Pediatrics 2015, 135, e598–e606. [Google Scholar] [CrossRef]
- Lax, S.; Smith, D.P.; Hampton-Marcell, J.; Owens, S.M.; Handley, K.M.; Scott, N.M.; Gibbons, S.M.; Larsen, P.; Shogan, B.D.; Weiss, S. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014, 345, 1048–1052. [Google Scholar] [CrossRef]
- Adams, R.I.; Sylvain, I.; Spilak, M.P.; Taylor, J.W.; Waring, M.S.; Mendell, M.J. Fungal signature of moisture damage in buildings: Identification by targeted and untargeted approaches with mycobiome data. Appl. Environ. Microbiol. 2020, 86, e01047-20. [Google Scholar] [CrossRef]
- Cabral, J.P. Can we use indoor fungi as bioindicators of indoor air quality? Historical perspectives and open questions. Sci. Total Environ. 2010, 408, 4285–4295. [Google Scholar] [CrossRef]
- Haas, D.; Habib, J.; Galler, H.; Buzina, W.; Schlacher, R.; Marth, E.; Reinthaler, F. Assessment of indoor air in austrian apartments with and without visible mold growth. Atmos. Environ. 2007, 41, 5192–5201. [Google Scholar] [CrossRef]
- EPA Ten Things You Should Know About Mold. Available online: https://www.epa.gov/mold/ten-things-you-should-know-about-mold (accessed on 28 September 2022).
- Morey, P.; Andrew, M.; Ligman, B.; Jarvis, J. Hidden Mold Sometimes Enters the Indoor Air. In Proceedings of the 9th International Conference on Indoor Air Quality and Climate. Monterey, Calif.; Levine, H., Bendy, G., Eds.; International Academy of Indoor Air Sciences: Monterey, CA, USA, 2002; pp. 455–460. [Google Scholar]
- Gravesen, S.; Nielsen, P.A.; Iversen, R.; Nielsen, K.F. Microfungal contamination of damp buildings—Examples of risk constructions and risk materials. Environ. Health Perspect. 1999, 107, 505–508. [Google Scholar] [CrossRef]
- Garrett, M.H.; Rayment, P.R.; Hooper, M.A.; Abramson, M.J.; Hooper, B.M. Indoor airborne fungal spores, house dampness and associations with environmental factors and respiratory health in children. Clin. Exp. Allergy 1998, 28, 459–467. [Google Scholar] [CrossRef]
- Miller, J.D.; Haisley, P.D.; Reinhardt, J.H. Air sampling results in relation to extent of fungal colonization of building materials in some water-damaged buildings. Indoor Air 2000, 10, 146–151. [Google Scholar] [CrossRef][Green Version]
- Mistry, H.; Soberanis, H.M.A.; Kyyaly, M.A.; Azim, A.; Barber, C.; Knight, D.; Newell, C.; Haitchi, H.M.; Wilkinson, T.; Howarth, P. The clinical implications of Aspergillus fumigatus sensitization in difficult-to-treat asthma patients. J. Allergy Clin. Immunol. Pract. 2021, 9, 4254–4267.e4210. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Wu, Z.; Wei, N.; Huang, H.; Zheng, P.; Liu, Y.; Sun, B. Molecular allergen sensitization of Aspergillus fumigatus between allergic bronchopulmonary aspergillosis and a fumigatus-sensitized asthma in guangzhou, southern china. J. Clin. Lab. Anal. 2020, 34, e23448. [Google Scholar] [CrossRef]
- Druey, K.M.; McCullough, M.; Krishnan, R. Aspergillus fumigatus protease alkaline protease 1 (alp1): A new therapeutic target for fungal asthma. J. Fungi 2020, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.; Hunt, E.B.; Ward, C.; Lapthorne, S.; Eustace, J.A.; Fanning, L.J.; Plant, B.J.; O’Byrne, P.M.; MacSharry, J.A.; Murphy, D.M. The presence of Aspergillus fumigatus in asthmatic airways is not clearly related to clinical disease severity. Allergy 2020, 75, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Namvar, S.; Labram, B.; Rowley, J.; Herrick, S. Aspergillus fumigatus—Host interactions mediating airway wall remodelling in asthma. J. Fungi 2022, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Godish, T.; Godish, D. Inter-laboratory variability in total mould particle counts based on co-located, concurrently collected total airborne samples. In Proceedings of the Indoor Environmental Quality, Problems, Research and Solutions, Durham, NC, USA, 17–19 July 2006. [Google Scholar]
- Robertson, L.D.; Brandys, R. A multi-laboratory comparative study of spore trap analyses. Mycologia 2011, 103, 226–231. [Google Scholar] [CrossRef]
- Vesper, S. Traditional mould analysis compared to a DNA-based method of mould analysis. Crit. Rev. Microbiol. 2011, 37, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmann, M.W.; Coronado, G.; Thompson, B.; Griffith, W.C.; Hanson, J.D.; Vesper, S.; Faustman, E.M. Utilizing pyrosequencing and quantitative pcr to characterize fungal populations among house dust samples. J. Environ. Monit. 2012, 14, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Bhunia, A.K. Mammalian cell-based biosensors for pathogens and toxins. Trends Biotechnol. 2009, 27, 179–188. [Google Scholar] [CrossRef]
- Bahram, M.; Netherway, T.; Frioux, C.; Ferretti, P.; Coelho, L.P.; Geisen, S.; Bork, P.; Hildebrand, F. Metagenomic assessment of the global diversity and distribution of bacteria and fungi. Environ. Microbiol. 2021, 23, 316–326. [Google Scholar] [CrossRef]
- Dunn, R.R.; Fierer, N.; Henley, J.B.; Leff, J.W.; Menninger, H.L. Home life: Factors structuring the bacterial diversity found within and between homes. PLoS ONE 2013, 8, e64133. [Google Scholar] [CrossRef]
- Tringe, S.G.; Zhang, T.; Liu, X.; Yu, Y.; Lee, W.H.; Yap, J.; Yao, F.; Suan, S.T.; Ing, S.K.; Haynes, M. The airborne metagenome in an indoor urban environment. PLoS ONE 2008, 3, e1862. [Google Scholar] [CrossRef] [PubMed]
- Ashley, P.; Dewalt, G.; Hamilton, R.; Jones, J.; Pinzer, E. Vacuum Dust Sample Collection Protocol for Allergens. HUD Office of Healthy Homes and Lead Hazard Control: 2008. Available online: https://www.hud.gov/sites/documents/DOC_12539.PDF (accessed on 9 May 2023).
- Mukherjee, N.; Bartelli, D.; Patra, C.; Chauhan, B.V.; Dowd, S.E.; Banerjee, P. Microbial diversity of source and point-of-use water in rural haiti–a pyrosequencing-based metagenomic survey. PLoS ONE 2016, 11, e0167353. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16s rdna bacterial tag-encoded flx amplicon pyrosequencing (btefap). BMC Microbiol. 2008, 8, 125. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Mukherjee, N.; Dowd, S.E.; Wise, A.; Kedia, S.; Vohra, V.; Banerjee, P. Diversity of bacterial communities of fitness center surfaces in a us metropolitan area. Int. J. Environ. Res. Public Health 2014, 11, 12544–12561. [Google Scholar] [CrossRef]
- Swanson, K.S.; Dowd, S.E.; Suchodolski, J.S.; Middelbos, I.S.; Vester, B.M.; Barry, K.A.; Nelson, K.E.; Torralba, M.; Henrissat, B.; Coutinho, P.M. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J. 2011, 5, 639–649. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rrna analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using microbiomeanalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Vesper, S.J.; McKinstry, C.; Haugland, R.A.; Iossifova, Y.; Lemasters, G.; Levin, L.; Khurana Hershey, G.K.; Villareal, M.; Bernstein, D.I.; Lockey, J.; et al. Relative moldiness index as predictor of childhood respiratory illness. J. Expo. Sci. Env. Epidemiol. 2007, 17, 88–94. [Google Scholar] [CrossRef]
- Reponen, T.; Lockey, J.; Bernstein, D.I.; Vesper, S.J.; Levin, L.; Khurana Hershey, G.K.; Zheng, S.; Ryan, P.; Grinshpun, S.A.; Villareal, M.; et al. Infant origins of childhood asthma associated with specific molds. J. Allergy Clin. Immunol. 2012, 130, 639–644.e635. [Google Scholar] [CrossRef]
- Vesper, S.; McKinstry, C.; Haugland, R.; Wymer, L.; Bradham, K.; Ashley, P.; Cox, D.; Dewalt, G.; Friedman, W. Development of an environmental relative moldiness index for us homes. J. Occup. Env. Med. 2007, 49, 829–833. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Johnson, T.A.; Su, J.-Q.; Qiao, M.; Guo, G.-X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef]
- Haugland, R.; Vesper, S. Method of identifying and quantifying specific fungi and bacteria. U.S. Patent 6,387,652, 14 May 2002. [Google Scholar]
- Haugland, R.A.; Varma, M.; Wymer, L.J.; Vesper, S.J. Quantitative pcr analysis of selected Aspergillus, Penicillium and Paecilomyces species. Syst. Appl. Microbiol. 2004, 27, 198–210. [Google Scholar] [CrossRef]
- Dixon, P. Vegan, a package of r functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot: Using the Grammar of Graphics with r; Springer: New York, NY, USA, 2009. [Google Scholar]
- Knief, C.; Ramette, A.; Frances, L.; Alonso-Blanco, C.; Vorholt, J.A. Site and plant species are important determinants of the methylobacterium community composition in the plant phyllosphere. ISME J. 2010, 4, 719–728. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar]
- Hammer, O.; Harper, D.A.; Ryan, P.D. Palaeontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4. Available online: http://palaeo-electronica.org/2001_1/past/issue1_01.htm (accessed on 28 September 2022).
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. Tbtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Rocchi, S.; Reboux, G.; Scherer, E.; Laboissière, A.; Zaros, C.; Rouzet, A.; Valot, B.; Khan, S.; Dufourg, M.-N.; Leynaert, B. Indoor microbiome: Quantification of exposure and association with geographical location, meteorological factors, and land use in france. Microorganisms 2020, 8, 341. [Google Scholar] [CrossRef]
- Coombs, K.; Taft, D.; Ward, D.V.; Green, B.J.; Chew, G.L.; Shamsaei, B.; Meller, J.; Indugula, R.; Reponen, T. Variability of indoor fungal microbiome of green and non-green low-income homes in cincinnati, ohio. Sci. Total Env. 2018, 610–611, 212–218. [Google Scholar] [CrossRef]
- Chew, G.L.; Rogers, C.; Burge, H.A.; Muilenberg, M.L.; Gold, D.R. Dustborne and airborne fungal propagules represent a different spectrum of fungi with differing relations to home characteristics. Allergy 2003, 58, 13–20. [Google Scholar] [CrossRef]
- Coombs, K.C.; Chew, G.L.; Schaffer, C.; Ryan, P.H.; Brokamp, C.; Grinshpun, S.A.; Adamkiewicz, G.; Chillrud, S.; Hedman, C.; Colton, M.; et al. Indoor air quality in green-renovated vs. Non-green low-income homes of children living in a temperate region of us (ohio). Sci. Total Environ. 2016, 554–555, 178–185. [Google Scholar] [CrossRef]
- Shin, E.K.; Shaban-Nejad, A. Urban decay and pediatric asthma prevalence in memphis, tennessee: Urban data integration for efficient population health surveillance. IEEE Access 2018, 6, 46281–46289. [Google Scholar] [CrossRef]
- Shin, E.K.; Mahajan, R.; Akbilgic, O.; Shaban-Nejad, A. Sociomarkers and biomarkers: Predictive modeling in identifying pediatric asthma patients at risk of hospital revisits. NPJ Digit. Med. 2018, 1, 50. [Google Scholar] [CrossRef]
- Lokida, D.; Hadi, U.; Lau, C.-Y.; Kosasih, H.; Liang, C.J.; Rusli, M.; Sudarmono, P.; Lukman, N.; Laras, K.; Asdie, R.H. Underdiagnoses of rickettsia in patients hospitalized with acute fever in indonesia: Observational study results. BMC Infect. Dis. 2020, 20, 364. [Google Scholar] [CrossRef]
- Rintala, H.; Pitkäranta, M.; Toivola, M.; Paulin, L.; Nevalainen, A. Diversity and seasonal dynamics of bacterial community in indoor environment. BMC Microbiol. 2008, 8, 56. [Google Scholar] [CrossRef]
- Bouillard, L.; Michel, O.; Dramaix, M.; Devleeschouwer, M. Bacterial contamination of indoor air, surfaces, and settled dust, and related dust endotoxin concentrations in healthy office buildings. Ann. Agric. Environ. Med. 2005, 12, 187–192. [Google Scholar]
- Lee, L.; Tin, S.; Kelley, S.T. Culture-independent analysis of bacterial diversity in a child-care facility. BMC Microbiol. 2007, 7, 27. [Google Scholar] [CrossRef]
- Abler, S.W. Ecology and Taxonomy of Leptosphaerulina spp. Associated with Turfgrasses in the United States. Ph.D. Thesis, Virginia Tech, Blacksburg, VA, USA, 2003. [Google Scholar]
- Victoria, A.; Furtado, B.; Holz, M.; Romero-Arenas, O.; Dallagnol, L. First report of leptosphaerulina leaf spot caused by leptosphaerulina trifolii on trifolium repens in brazil. Plant Dis. 2020, 104, 972. [Google Scholar] [CrossRef]
- Roux, C. Leptosphaerulina chartarum sp. Nov., the teleomorph of Pithomyces chartarum. Trans. Br. Mycol. Soc. 1986, 86, 319–323. [Google Scholar] [CrossRef]
- Pakpour, S.; Li, D.-W.; Klironomos, J. Relationships of fungal spore concentrations in the air and meteorological factors. Fungal Ecol. 2015, 13, 130–134. [Google Scholar] [CrossRef]
- Pitkäranta, M.; Meklin, T.; Hyvärinen, A.; Nevalainen, A.; Paulin, L.; Auvinen, P.; Lignell, U.; Rintala, H. Molecular profiling of fungal communities in moisture damaged buildings before and after remediation—A comparison of culture-dependent and culture-independent methods. BMC Microbiol. 2011, 11, 235. [Google Scholar] [CrossRef]
- Salo, P.M.; Arbes, S.J., Jr.; Sever, M.; Jaramillo, R.; Cohn, R.D.; London, S.J.; Zeldin, D.C. Exposure to alternaria alternata in us homes is associated with asthma symptoms. J. Allergy Clin. Immunol. 2006, 118, 892–898. [Google Scholar] [CrossRef]
- Bush, R.K.; Prochnau, J.J. Alternaria-induced asthma. J. Allergy Clin. Immunol. 2004, 113, 227–234. [Google Scholar] [CrossRef]
- Causton, B.; Pardo-Saganta, A.; Gillis, J.; Discipio, K.; Kooistra, T.; Rajagopal, J.; Xavier, R.J.; Cho, J.L.; Medoff, B.D. Carma3 mediates allergic lung inflammation in response to alternaria alternata. Am. J. Respir. Cell Mol. Biol. 2018, 59, 684–694. [Google Scholar] [CrossRef]
- Piecková, E. In vitro toxicity of indoor chaetomium kunze ex fr. Ann. Agric. Environ. Med. 2003, 10, 9–14. [Google Scholar]
- Abbott, S.P.; Sigler, L.; McAleer, R.; McGough, D.A.; Rinaldi, M.; Mizell, G. Fatal cerebral mycoses caused by the ascomycete chaetomium strumarium. J. Clin. Microbiol. 1995, 33, 2692–2698. [Google Scholar] [CrossRef]
- Serena, C.; Ortoneda, M.; Capilla, J.; Pastor, F.J.; Sutton, D.A.; Rinaldi, M.G.; Guarro, J. In vitro activities of new antifungal agents against Chaetomium spp. And inoculum standardization. Antimicrob. Agents Chemother. 2003, 47, 3161–3164. [Google Scholar] [CrossRef]
- Salo, J.M.; Kedves, O.; Mikkola, R.; Kredics, L.; Andersson, M.A.; Kurnitski, J.; Salonen, H. Detection of Chaetomium globosum, ch. Cochliodes and ch. Rectangulare during the diversity tracking of mycotoxin-producing chaetomium-like isolates obtained in buildings in finland. Toxins 2020, 12, 443. [Google Scholar] [CrossRef]
- Andersen, B.; Frisvad, J.C.; Dunn, R.R.; Thrane, U. A pilot study on baseline fungi and moisture indicator fungi in danish homes. J. Fungi 2021, 7, 71. [Google Scholar] [CrossRef]
- Omebeyinje, M.H.; Adeluyi, A.; Mitra, C.; Chakraborty, P.; Gandee, G.M.; Patel, N.; Verghese, B.; Farrance, C.E.; Hull, M.; Basu, P.; et al. Increased prevalence of indoor Aspergillus and Penicillium species is associated with indoor flooding and coastal proximity: A case study of 28 moldy buildings. Env. Sci. Process Impacts 2021, 23, 1681–1687. [Google Scholar] [CrossRef]
- Flores, G.E.; Bates, S.T.; Caporaso, J.G.; Lauber, C.L.; Leff, J.W.; Knight, R.; Fierer, N. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 2013, 15, 588–596. [Google Scholar] [CrossRef]
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. The diversity and distribution of fungi on residential surfaces. PLoS ONE 2013, 8, e78866. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. Microbiomes of the Built Environment: A Research Agenda for Indoor Microbiology, Human Health, and Buildings; The National Academies Press: Washington, DC, USA, 2017. [Google Scholar]
- Cortes-Ramirez, J.; Wilches-Vega, J.D.; Paris-Pineda, O.M.; Rod, J.; Ayurzana, L.; Sly, P.D. Environmental risk factors associated with respiratory diseases in children with socioeconomic disadvantage. Heliyon 2021, 7, e06820. [Google Scholar] [CrossRef]
- Kumar, R.K.; Herbert, C.; Foster, P.S. Mouse models of acute exacerbations of allergic asthma. Respirology 2016, 21, 842–849. [Google Scholar] [CrossRef]
- Gabriel, M.F.; Postigo, I.; Tomaz, C.T.; Martínez, J. Alternaria alternata allergens: Markers of exposure, phylogeny and risk of fungi-induced respiratory allergy. Environ. Int. 2016, 89, 71–80. [Google Scholar] [CrossRef]
- Kim, D.M.; Lee, M.H.; Suh, M.K.; Ha, G.Y.; Kim, H.; Choi, J.S. Onychomycosis caused by Chaetomium globosum. Ann. Dermatol. 2013, 25, 232–236. [Google Scholar] [CrossRef]
- Lewinska, A.M.; Hoof, J.B.; Peuhkuri, R.H.; Rode, C.; Lilje, O.; Foley, M.; Trimby, P.; Andersen, B. Visualization of the structural changes in plywood and gypsum board during the growth of Chaetomium globosum and Stachybotrys chartarum. J. Microbiol. Methods 2016, 129, 28–38. [Google Scholar] [CrossRef]
- Andersen, B.; Dosen, I.; Lewinska, A.M.; Nielsen, K.F. Pre-contamination of new gypsum wallboard with potentially harmful fungal species. Indoor Air 2017, 27, 6–12. [Google Scholar] [CrossRef]
- Ladd, T.B.; Johnson, J.A., Jr.; Mumaw, C.L.; Greve, H.J.; Xuei, X.; Simpson, E.; Barnes, M.A.; Green, B.J.; Croston, T.L.; Ahmed, C. Aspergillus versicolor inhalation triggers neuroimmune, glial, and neuropeptide transcriptional changes. ASN Neuro 2021, 13. [Google Scholar] [CrossRef]
- Hodgson, M.J.; Morey, P.; Leung, W.-Y.; Morrow, L.; Miller, D.; Jarvis, B.B.; Robbins, H.; Halsey, J.F.; Storey, E. Building-associated pulmonary disease from exposure to Stachybotrys chartarum and Aspergillus versicolor. J. Occup. Environ. Med. 1998, 40, 241–249. [Google Scholar] [CrossRef]
- Reponen, T.; Singh, U.; Schaffer, C.; Vesper, S.; Johansson, E.; Adhikari, A.; Grinshpun, S.A.; Indugula, R.; Ryan, P.; Levin, L. Visually observed mold and moldy odor versus quantitatively measured microbial exposure in homes. Sci. Total Environ. 2010, 408, 5565–5574. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| House Type | Correlation Between | Correlation Coefficient, rs | Correlation | p Value |
|---|---|---|---|---|
| Houses with no visible mold (HNM) | OTU: PM 1.0 | 0.22 | Positive | 0.53 |
| OTU: PM 2.5 | 0.22 | Positive | 0.53 | |
| OTU: PM 10.0 | 0.23 | Positive | 0.51 | |
| OTU: CO2 level | 0.35 | Positive | 0.38 | |
| OTU: Humidity | 0.28 | Positive | 0.49 | |
| OUT: Temperature | 0.38 | Positive | 0.27 | |
| Houses with visible mold (HVM) | OTU: PM 1.0 | −0.05 | Negative | 0.87 |
| OTU: PM 2.5 | −0.05 | Negative | 0.87 | |
| OTU: PM 10.0 | 0.01 | Positive | 0.96 | |
| OTU: CO2 level | −0.28 | Negative | 0.42 | |
| OTU: Humidity | −0.03 | Negative | 0.93 | |
| OTU: Temperature | −0.21 | Negative | 0.55 |
| House Type | Correlation Between | Correlation Coefficient, rs | Correlation | p Value |
|---|---|---|---|---|
| Houses with no visible mold (HNM) | OTU: PM 1.0 | 0.62 | Positive | 0.05 |
| OTU: PM 2.5 | 0.62 | Positive | 0.05 | |
| OTU: PM 10.0 | 0.46 | Positive | 0.17 | |
| OTU: CO2 level | 0.23 | Positive | 0.57 | |
| OTU: Humidity | 0.30 | Positive | 0.45 | |
| OTU: Temperature | −0.22 | Negative | 0.52 | |
| Houses with visible mold (HVM) | OTU: PM 1.0 | −0.48 | Negative | 0.17 |
| OTU: PM 2.5 | −0.48 | Negative | 0.17 | |
| OTU: PM 10.0 | −0.45 | Negative | 0.19 | |
| OTU: CO2 level | −0.45 | Negative | 0.18 | |
| OTU: Humidity | 0.10 | Positive | 0.77 | |
| OTU: Temperature | 0.11 | Positive | 0.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, B.V.; Higgins Jones, D.; Banerjee, G.; Agrawal, S.; Sulaiman, I.M.; Jia, C.; Banerjee, P. Indoor Bacterial and Fungal Burden in “Moldy” versus “Non-Moldy” Homes: A Case Study Employing Advanced Sequencing Techniques in a US Metropolitan Area. Pathogens 2023, 12, 1006. https://doi.org/10.3390/pathogens12081006
Chauhan BV, Higgins Jones D, Banerjee G, Agrawal S, Sulaiman IM, Jia C, Banerjee P. Indoor Bacterial and Fungal Burden in “Moldy” versus “Non-Moldy” Homes: A Case Study Employing Advanced Sequencing Techniques in a US Metropolitan Area. Pathogens. 2023; 12(8):1006. https://doi.org/10.3390/pathogens12081006
Chicago/Turabian StyleChauhan, Bhavin V., Daleniece Higgins Jones, Goutam Banerjee, Saumya Agrawal, Irshad M. Sulaiman, Chunrong Jia, and Pratik Banerjee. 2023. "Indoor Bacterial and Fungal Burden in “Moldy” versus “Non-Moldy” Homes: A Case Study Employing Advanced Sequencing Techniques in a US Metropolitan Area" Pathogens 12, no. 8: 1006. https://doi.org/10.3390/pathogens12081006
APA StyleChauhan, B. V., Higgins Jones, D., Banerjee, G., Agrawal, S., Sulaiman, I. M., Jia, C., & Banerjee, P. (2023). Indoor Bacterial and Fungal Burden in “Moldy” versus “Non-Moldy” Homes: A Case Study Employing Advanced Sequencing Techniques in a US Metropolitan Area. Pathogens, 12(8), 1006. https://doi.org/10.3390/pathogens12081006