

A Rose by Any Other Name: The Long Intricate History of Localized Aggressive Periodontitis



Abstract
1. Introduction: Importance of Recognizing Subtypes of Periodontitis for Scientific Discovery
2. Influence of Consensus Conference and Key Microbiological Influences on Disease Classification
- (A)
- WWCC impact on Disease Classification.
- (B)
- Support for Maintaining LAgP as a Disease entity.
- (C)
- Influence of WWCC decision on LAgP publications.
3. Benefits of Precise Disease Definitions
4. Brief History of Landmark Experiments That Support Microbiological Associations with Dental Diseases
- (A)
- Seminal Discoveries of Microbes and Dental Diseases.
- (B)
- Microbiology/A. actinomycetemcomitans and LAgP.
- (C)
- Bradford–Hill Guidelines for Determining Causation: Association of A. actinomycetemcomitans in a Microbial Consortium with LAgP.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hill Criteria | Example | Feasibility Yes/No? | Impact of Study and Reference |
|---|---|---|---|
| 1. Temporal relationship | Exposure to agent precedes outcome | Yes | Longitudinal; healthy controls; age approp Aa; [98,99] Longitudinal; health controls age approp Aa + consort; [29,100] |
| 2. Strength of Association | Size of association determined statistically | Yes | Show stats Aa; [98] Show stats Aa + consort; [29,100] |
| 3. Dose-Response | ^exposure > ^response | Yes | Measure consort vs. Aa alone; [29,100] |
| 4. Consistency | Experiments reproduced | Yes | Show consort X-sect; [101,102] Show consort longitude; [29,100] |
| 5. Plausibility | Assoc agrees with pathobiological explanations | Yes | Cdt has impact; [103] Ltx has impact; [95] Consortia passed from mother with disease to Child: local debridement improves inflammation, but consort remains; [104]. Consort metabolomics; [94] |
| 6. Experimental evidence | Disease altered By intervention | Yes | Tetracycline admin reduces disease; [105] Tetracycline eliminates Aa and reduces disease; [106] Amox/Metra reduces disease, no antibiotic, no improvement; [107] |
| 7. Alternative explanation | Rule out other explanations | ? Open ? Open | [29,98,100] |
| 8. Specificity | Cause produces effect | Yes | Flp and no disease; [108] Ltx and more bone loss; [109] Pga B is modified, and disease is reduced; [110] |
| 9. Coherence | Theory consistent with Existing knowledge | Yes | Ltx and infections; [111] Cdt and infections; [112] Metabolomics and consortia; [94] |
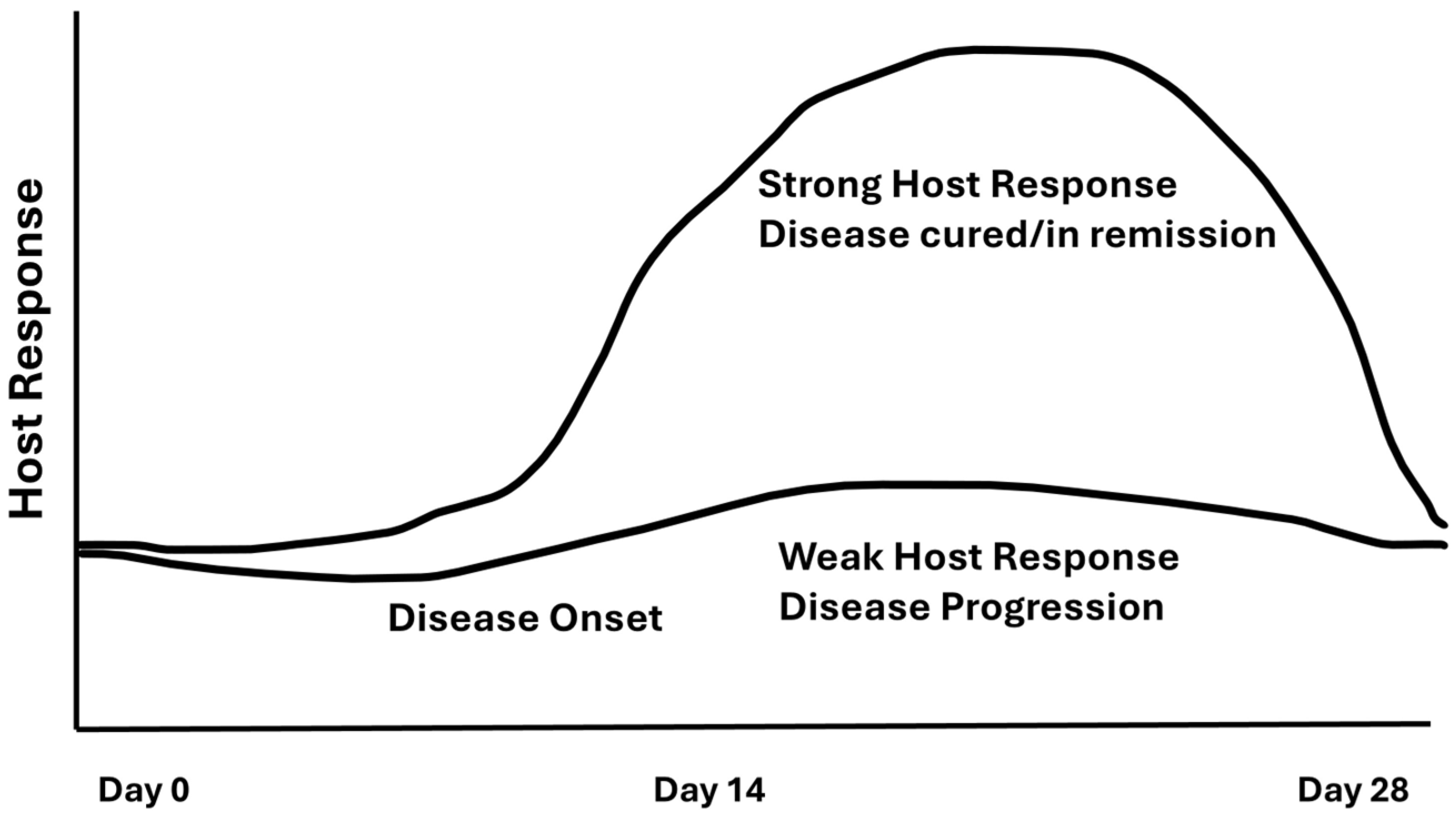
5. Damage/Response Framework and LAgP
- (A)
- Overview.
- (B)
- Damage.
- (C)
- Response.
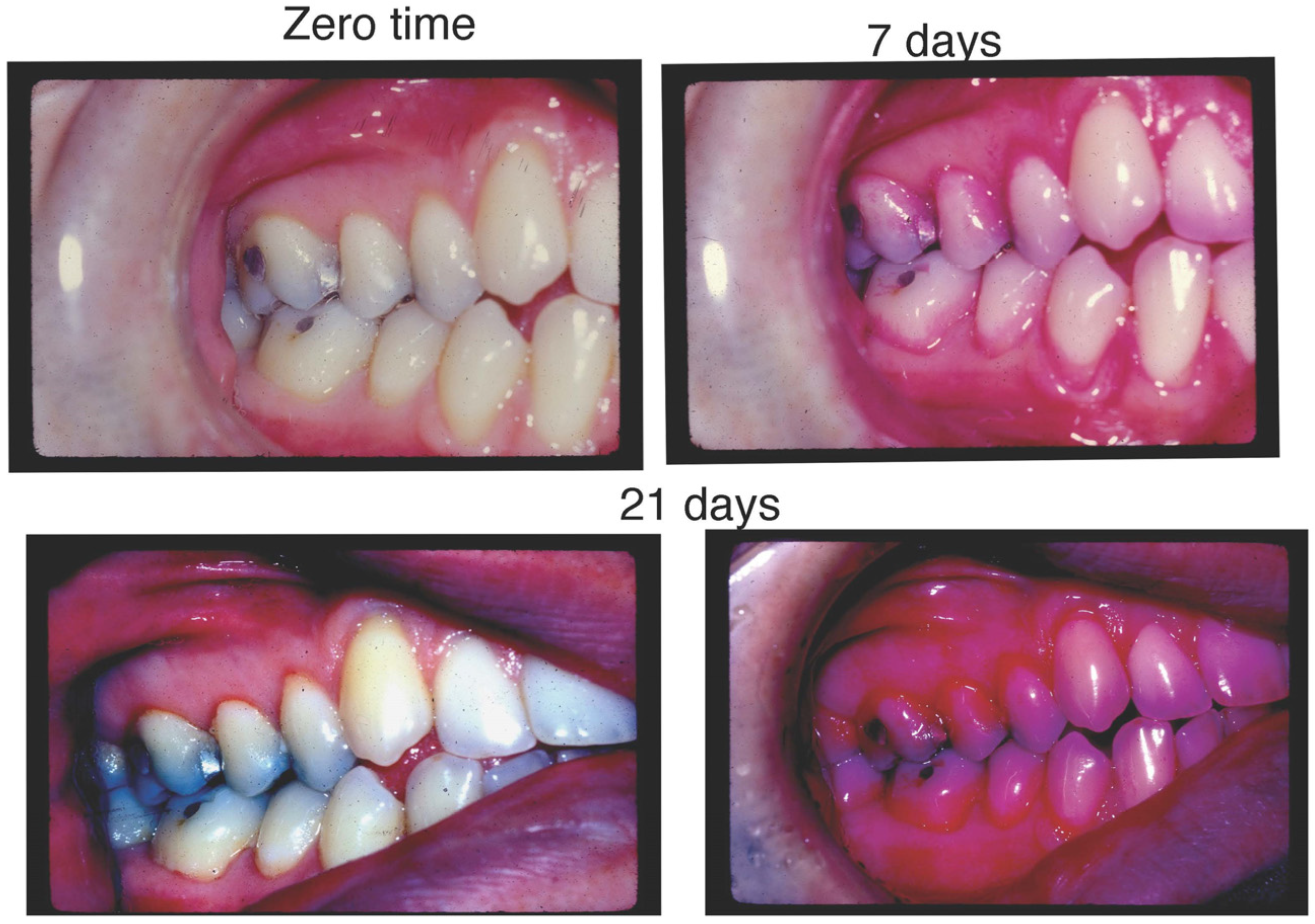
6. Distinct Characteristics of LAgP and an Alternative Approach for Assessing Treatment Success
- (A)
- Distinct Characteristics of LAgP: An Illustrative Case.
- (B)
- Another way of Assessing Periodontitis Treatment Success.
7. Conclusions: Future Challenges/Recommendations for the Clinical and Research Community
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S173–S182. [Google Scholar] [CrossRef] [PubMed]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J. Clin. Periodontol. 2018, 45 (Suppl. S20), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Periodontal diseases: Diagnosis. Ann. Periodontol. 1996, 1, 37–215. [Google Scholar] [CrossRef] [PubMed]
- Greene, F.L. Cancer staging in outcomes assessment. J. Surg. Oncol. 2014, 110, 616–620. [Google Scholar] [CrossRef]
- Park, B.K.; Schneider, J.; Suh, Y.J. Survival analysis for patients with metachronous contralateral breast cancer: Insights from a retrospective study. Oncol. Lett. 2024, 28, 390. [Google Scholar] [CrossRef]
- Baer, P.N. The case for periodontosis as a clinical entity. J. Periodontol. 1971, 42, 516–520. [Google Scholar] [CrossRef]
- Marazita, M.L.; Burmeister, J.A.; Gunsolley, J.C.; Koertge, T.E.; Lake, K.; Schenkein, H.A. Evidence for autosomal dominant inheritance and race-specific heterogeneity in early-onset periodontitis. J. Periodontol. 1994, 65, 623–630. [Google Scholar] [CrossRef]
- Loe, H.; Brown, L.J. Early onset periodontitis in the United States of America. J. Periodontol. 1991, 62, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Kaplan, J.B.; Kachlany, S.C.; Schreiner, H.C. How we got attached to Actinobacillus actinomycetemcomitans: A model for infectious diseases. Periodontol. 2000 2006, 42, 114–157. [Google Scholar] [CrossRef]
- Fine, D.H.; Cohen, D.W.; Bimstein, E.; Bruckmann, C. A ninety-year history of periodontosis: The legacy of Professor Bernhard Gottlieb. J. Periodontol. 2015, 86, 1–6. [Google Scholar] [CrossRef]
- Zambon, J.J. Authors’ response. J. Am. Dent. Assoc. 2020, 151, 160–161. [Google Scholar] [CrossRef]
- de Carvalho, F.M.; Tinoco, E.M.; Govil, M.; Marazita, M.L.; Vieira, A.R. Aggressive periodontitis is likely influenced by a few small effect genes. J. Clin. Periodontol. 2009, 36, 468–473. [Google Scholar] [CrossRef]
- Fine, D.H.; Armitage, G.C.; Genco, R.J.; Griffen, A.L.; Diehl, S.R. Unique etiologic, demographic, and pathologic characteristics of localized aggressive periodontitis support classification as a distinct subcategory of periodontitis. J. Am. Dent. Assoc. 2019, 150, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Pallos, D.; Acevedo, A.C.; Mestrinho, H.D.; Cordeiro, I.; Hart, T.C. Novel cathepsin C mutation in a Brazilian family with Papillon-Lefevre syndrome: Case report and mutation update. J. Dent. Child. 2010, 77, 36–41. [Google Scholar]
- Diehl, S.R.; Wu, T.; Michalowicz, B.S.; Brooks, C.N.; Califano, J.V.; Burmeister, J.A.; Schenkein, H.A. Quantitative measures of aggressive periodontitis show substantial heritability and consistency with traditional diagnoses. J. Periodontol. 2005, 76, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W. Development of orphan drugs for rare diseases. Clin. Exp. Pediatr. 2024, 67, 315–327. [Google Scholar] [CrossRef]
- Wade, D.N.; Kerns, D.G. Acute necrotizing ulcerative gingivitis-periodontitis: A literature review. Mil. Med. 1998, 163, 337–342. [Google Scholar] [CrossRef]
- Albandar, J.M. Disparities and social determinants of periodontal diseases. Periodontol. 2000 2024. [Google Scholar] [CrossRef]
- Botelho, J.; Machado, V.; Proenca, L.; Mendes, J.J. The 2018 periodontitis case definition improves accuracy performance of full-mouth partial diagnostic protocols. Sci. Rep. 2020, 10, 7093. [Google Scholar] [CrossRef]
- Fredman, G.; Oh, S.F.; Ayilavarapu, S.; Hasturk, H.; Serhan, C.N.; Van Dyke, T.E. Impaired phagocytosis in localized aggressive periodontitis: Rescue by Resolvin E1. PLoS ONE 2011, 6, e24422. [Google Scholar] [CrossRef]
- Fine, D.H.; Patil, A.G.; Loos, B.G. Classification and diagnosis of aggressive periodontitis. J. Periodontol. 2018, 89 (Suppl. S1), S103–S119. [Google Scholar] [CrossRef] [PubMed]
- Endersby, J. Lumpers and splitters: Darwin, Hooker, and the search for order. Science 2009, 326, 1496–1499. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer; Scribner: New York, NY, USA, 2010. [Google Scholar]
- Denoix, P.F. Présentation d’une nomenclature classification des cancers basee sur un atlas [Nomenclature and classification of cancers based on an atlas]. Acta Unio Int. Contra Cancrum 1953, 9, 769–771. [Google Scholar] [PubMed]
- Snyderman, R. Personalized health care: From theory to practice. Biotechnol. J. 2012, 7, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Albandar, J.M. Aggressive periodontitis: Case definition and diagnostic criteria. Periodontol. 2000 2014, 65, 13–26. [Google Scholar] [CrossRef]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol. 2000 2014, 64, 57–80. [Google Scholar] [CrossRef]
- Billings, M.; Holtfreter, B.; Papapanou, P.N.; Mitnik, G.L.; Kocher, T.; Dye, B.A. Age-dependent distribution of periodontitis in two countries: Findings from NHANES 2009 to 2014 and SHIP-TREND 2008 to 2012. J. Periodontol. 2018, 89 (Suppl. S1), S140–S158. [Google Scholar] [CrossRef]
- Fine, D.H.; Markowitz, K.; Fairlie, K.; Tischio-Bereski, D.; Ferrendiz, J.; Furgang, D.; Paster, B.J.; Dewhirst, F.E. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J. Clin. Microbiol. 2013, 51, 2850–2861. [Google Scholar] [CrossRef]
- Needleman, I.; Garcia, R.; Gkranias, N.; Kirkwood, K.L.; Kocher, T.; Iorio, A.D.; Moreno, F.; Petrie, A. Mean annual attachment, bone level, and tooth loss: A systematic review. J. Periodontol. 2018, 89 (Suppl. S1), S120–S139. [Google Scholar] [CrossRef]
- Leow, N.M.; Moreno, F.; Marletta, D.; Hussain, S.B.; Buti, J.; Almond, N.; Needleman, I. Recurrence and progression of periodontitis and methods of management in long-term care: A systematic review and meta-analysis. J. Clin. Periodontol. 2022, 49 (Suppl. S24), 291–313. [Google Scholar] [CrossRef]
- Fine, D.H.; Armitage, G.C.; Griffen, A.L.; Diehl, S.R. Authors’ response. J. Am. Dent. Assoc. 2020, 151, 160. [Google Scholar] [CrossRef]
- Bik, E.M.; Long, C.D.; Armitage, G.C.; Loomer, P.; Emerson, J.; Mongodin, E.F.; Nelson, K.E.; Gill, S.R.; Fraser-Liggett, C.M.; Relman, D.A. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010, 4, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.M.; Relman, D.A. The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host Microbe 2017, 21, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta-catenin modulator Annexin A1. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Han, Y.W.; Redline, R.W.; Li, M.; Yin, L.; Hill, G.B.; McCormick, T.S. Fusobacterium nucleatum Induces Premature and Term Stillbirths in Pregnant Mice: Implication of Oral Bacteria in Preterm Birth. Infect. Immun. 2004, 72, 2272–2279. [Google Scholar] [CrossRef] [PubMed]
- Zambon, J.J. Periodontal diseases: Microbial factors. Ann Periodontol 1996, 1, 879–925. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- American Academy of Pediatric Dentistry Council on Clinical Affairs. Policy on use of a caries-risk assessment tool (CAT) for infants, children, and adolescents. Pediatr. Dent. 2005, 27, 25–27. [Google Scholar]
- Loesche, W.J. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 1986, 50, 353–380. [Google Scholar] [CrossRef]
- Marsh, P.D. Are dental diseases examples of ecological catastrophes? Microbiology 2003, 149, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Folayan, M.N.O.; Amalia, R.; Kemoli, A.; Sun, I.G.; Duangthip, D.; Abodunrin, O.; Virtanen, J.I.; Masumo, R.M.; Vukovic, A.; Al-Batayneh, O.B.; et al. Can the sustainable development goal 9 support an untreated early childhood caries elimination agenda? BMC Oral Health 2024, 24, 776. [Google Scholar] [CrossRef] [PubMed]
- Sheykholeslam, Z.; Buonocore, M.G. Bonding of resins to phosphoric acid-etched enamel surfaces of permanent and deciduous teeth. J. Dent. Res. 1972, 51, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, A.Y.; Geissler, K.H.; Dick, A.W.; Kranz, A.M. Clinician characteristics associated with fluoride varnish applications during well-child visits. Am. J. Manag. Care 2024, 30, e203–e209. [Google Scholar] [CrossRef]
- Lumsden, C.L.; Edelstein, B.L.; Basch, C.E.; Wolf, R.L.; Koch, P.A.; McKeague, I.; Leu, C.S.; Andrews, H. Protocol for a family-centered behavioral intervention to reduce early childhood caries: The MySmileBuddy program efficacy trial. BMC Oral Health 2021, 21, 246. [Google Scholar] [CrossRef]
- Lienhart, G.; Elsa, M.; Farge, P.; Schott, A.M.; Thivichon-Prince, B.; Chaneliere, M. Factors perceived by health professionals to be barriers or facilitators to caries prevention in children: A systematic review. BMC Oral Health 2023, 23, 767. [Google Scholar] [CrossRef]
- Woodward, C.; Fisher, M.A. Drug treatment of common STDs: Part II. Vaginal infections, pelvic inflammatory disease and genital warts. Am. Fam. Physician. 1999, 60, 1716–1722. [Google Scholar] [PubMed]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: An update. Indian J. Med. Res. 2013, 138, 303–316. [Google Scholar]
- Radu, O.; Pantanowitz, L. Kaposi sarcoma. Arch. Pathol. Lab. Med. 2013, 137, 289–294. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infect. Immun. 1999, 67, 3703–3713. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Microbiology: Ditch the term pathogen. Nature 2014, 516, 165–166. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. What is a pathogen? Ann. Med. 2002, 34, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.; Casadevall, A. The Damage-Response Framework as a Tool for the Physician-Scientist to Understand the Pathogenesis of Infectious Diseases. J. Infect. Dis. 2018, 218, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.A.; Casadevall, A. The meaning of microbial exposure, infection, colonisation, and disease in clinical practice. Lancet Infect. Dis. 2002, 2, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.D. The Micro-Organisms of the Human Mouth: The Local and General Diseases Which are Caused by Them; Classics of Dentistry Library: Sydney, Australia, 1890. [Google Scholar]
- Kritchevsky, B.; Seguin, P. The Pathogenesis and Treatment of Pyorrhea Alveolaris. Dent. Cosm. A Mon. Rec. Dent. Sci. 1918, 60, 781–784. [Google Scholar]
- Beckwith, T.D.; Williams, A.; Rose, E.T. The role of bacteria in pyorrhea. Med. J. Rec. 1929, 129, 333–336. [Google Scholar]
- Keyes, P.H.; Fitzgerald, R.J. Dental caries in the Syrian hamster. IX. Arch. Oral. Biol. 1962, 7, 267–277. [Google Scholar] [CrossRef]
- Loe, H.; Theilade, E.; Jensen, S.B. Experimental Gingivitis in Man. J. Periodontol. 1965, 36, 177–187. [Google Scholar] [CrossRef]
- Fine, D.H. Incorporating new technologies in periodontal diagnosis into training programs and patient care: A critical assessment and a plan for the future. J. Periodontol. 1992, 63, 383–393. [Google Scholar] [CrossRef]
- Listgarten, M.A. Periodontal probing: What does it mean? J. Clin. Periodontol. 1980, 7, 165–176. [Google Scholar] [CrossRef]
- Cugini, C.; Ramasubbu, N.; Tsiagbe, V.K.; Fine, D.H. Dysbiosis From a Microbial and Host Perspective Relative to Oral Health and Disease. Front. Microbiol. 2021, 12, 617485. [Google Scholar] [CrossRef] [PubMed]
- Page, R.C.; Schroeder, H.E. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab. Investig. 1976, 34, 235–249. [Google Scholar] [PubMed]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontol. 2000 2020, 83, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Lang, N.P.; Kiel, R.A.; Anderhalden, K. Clinical and microbiological effects of subgingival restorations with overhanging or clinically perfect margins. J. Clin. Periodontol. 1983, 10, 563–578. [Google Scholar] [CrossRef]
- Theilade, E.; Wright, W.H.; Jensen, S.B.; Loe, H. Experimental gingivitis in man. II. A longitudinal clinical and bacteriological investigation. J. Periodontal. Res. 1966, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bamashmous, S.; Kotsakis, G.A.; Kerns, K.A.; Leroux, B.G.; Zenobia, C.; Chen, D.; Trivedi, H.M.; McLean, J.S.; Darveau, R.P. Human variation in gingival inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2012578118. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; London, J. Adhere today, here tomorrow: Oral bacterial adherence. J. Bacteriol. 1993, 175, 3247–3252. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. What is a host? Incorporating the microbiota into the damage-response framework. Infect. Immun. 2015, 83, 2–7. [Google Scholar] [CrossRef]
- Loe, H.; Silness, J. Periodontal disease in pregancy. Prevalence and severity. Acta Odontol Scand 1963, 21, 533–551. [Google Scholar] [CrossRef]
- Armitage, G.C. Learned and unlearned concepts in periodontal diagnostics: A 50-year perspective. Periodontol. 2000 2013, 62, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.G.; Socransky, S.S.; Savitt, E.D.; Propas, D.A.; Crawford, A. Studies of the microbiology of periodontosis. J. Periodontol. 1976, 47, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. The predominant cultivable organisms in juvenile periodontitis. Scand. J. Dent. Res. 1976, 84, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Loesche, W.J. Chemotherapy of dental plaque infections. Oral. Sci. Rev. 1976, 9, 65–107. [Google Scholar] [PubMed]
- Loesche, W.J. Clinical and microbiological aspects of chemotherapeutic agents used according to the specific plaque hypothesis. J. Dent. Res. 1979, 58, 2404–2412. [Google Scholar] [CrossRef]
- Klinger, R. Untersuchungen uber menschliche aktinomycose. Zentralbl. Bakteriol. 1912, 62, 191–200. [Google Scholar]
- Marsh, P.D.; Zaura, E. Dental biofilm: Ecological interactions in health and disease. J. Clin. Periodontol. 2017, 44 (Suppl. S18), S12–S22. [Google Scholar] [CrossRef]
- Moore, W.E.; Moore, L.H.; Ranney, R.R.; Smibert, R.M.; Burmeister, J.A.; Schenkein, H.A. The microflora of periodontal sites showing active destructive progression. J. Clin. Periodontol. 1991, 18, 729–739. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal microbial ecology. Periodontol. 2000 2005, 38, 135–187. [Google Scholar] [CrossRef]
- Marsh, P.D. In Sickness and in Health-What Does the Oral Microbiome Mean to Us? An Ecological Perspective. Adv. Dent. Res. 2018, 29, 60–65. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Macdonald, J.B. Hemin and vitamin K compounds as required factors for the cultivation of certain strains of Bacteroides melaninogenicus. J. Bacteriol. 1960, 80, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, K.; Khandelwal, R.L.; Heinrich, S.E. Glycogen synthetic and degradative activities by Actinomyces viscosus and Actinomyces naeslundii of root surface caries and noncaries sites. Caries Res. 1988, 22, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.E.C.; Moore, L.V.H. The bacteria of periodontal disease. Periodontol. 2000 1994, 5, 66–77. [Google Scholar] [CrossRef]
- Schachtele, C.F.; Loken, A.E.; Schmitt, M.K. Use of specifically labeled sucrose for comparison of extracellular glucan and fructan metabolism by oral streptococci. Infect. Immun. 1972, 5, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Kachlany, S.C.; Fine, D.H.; Figurski, D.H. Secretion of RTX leukotoxin by Actinobacillus actinomycetemcomitans. Infect. Immun. 2000, 68, 6094–6100. [Google Scholar] [CrossRef]
- Pavloff, N.; Potempa, J.; Pike, R.N.; Prochazka, V.; Kiefer, M.C.; Travis, J.; Barr, P.J. Molecular cloning and structural characterization of the Arg-gingipain proteinase of Porphyromonas gingivalis. Biosynthesis as a proteinase-adhesin polyprotein. J. Biol. Chem. 1995, 270, 1007–1010. [Google Scholar] [CrossRef]
- Chu, L.; Bramanti, T.E.; Ebersole, J.L.; Holt, S.C. Hemolytic activity in the periodontopathogen Porphyromonas gingivalis: Kinetics of enzyme release and localization. Infect. Immun. 1991, 59, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; Macdonald, J.B. Degradation of collagenous substrates by Bacteroides melaninogenicus. J. Bacteriol. 1961, 81, 614–621. [Google Scholar] [CrossRef]
- Marsh, P.D. Microbial ecology of dental plaque and its significance in health and disease. Adv. Dent. Res. 1994, 8, 263–271. [Google Scholar] [CrossRef]
- Usman, M.; Hameed, Y.; Ahmad, M. Does human papillomavirus cause human colorectal cancer? Applying Bradford Hill criteria postulates. Ecancermedicalscience 2020, 14, 1107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wright, C.J.; Dingming, H.; Uriarte, S.M.; Lamont, R.J. Oral community interactions of Filifactor alocis in vitro. PLoS ONE 2013, 8, e76271. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Claesson, R.; Aberg, C.H.; Haubek, D.; Lindholm, M.; Jasim, S.; Oscarsson, J. Genetic Profiling of Aggragatibacter actinomycetemcomitans Serotype B Isolated from Periodontitis Patients Living in Sweden. Pathogens 2019, 8, 153. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Patel, M.; Socransky, S.S. Microbiological changes associated with four different periodontal therapies for the treatment of chronic periodontitis. Oral Microbiol. Immunol. 2008, 23, 148–157. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Microbial mechanisms in the pathogenesis of destructive periodontal diseases: A critical assessment. J. Periodontal. Res. 1991, 26, 195–212. [Google Scholar] [CrossRef]
- Haubek, D.; Ennibi, O.K.; Poulsen, K.; Vaeth, M.; Poulsen, S.; Kilian, M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: A prospective longitudinal cohort study. Lancet 2008, 371, 237–242. [Google Scholar] [CrossRef]
- Hoglund Aberg, C.; Kwamin, F.; Claesson, R.; Dahlen, G.; Johansson, A.; Haubek, D. Progression of attachment loss is strongly associated with presence of the JP2 genotype of Aggregatibacter actinomycetemcomitans: A prospective cohort study of a young adolescent population. J. Clin. Periodontol. 2014, 41, 232–241. [Google Scholar] [CrossRef]
- Razooqi, Z.; Tjellstrom, I.; Hoglund Aberg, C.; Kwamin, F.; Claesson, R.; Haubek, D.; Johansson, A.; Oscarsson, J. Association of Filifactor alocis and its RTX toxin gene ftxA with periodontal attachment loss, and in synergy with Aggregatibacter actinomycetemcomitans. Front. Cell. Infect. Microbiol. 2024, 14, 1376358. [Google Scholar] [CrossRef] [PubMed]
- Shaddox, L.M.; Huang, H.; Lin, T.; Hou, W.; Harrison, P.L.; Aukhil, I.; Walker, C.B.; Klepac-Ceraj, V.; Paster, B.J. Microbiological characterization in children with aggressive periodontitis. J. Dent. Res. 2012, 91, 927–933. [Google Scholar] [CrossRef]
- Koo, S.S.; Fernandes, J.G.; Li, L.; Huang, H.; Aukhil, I.; Harrison, P.; Diaz, P.I.; Shaddox, L.M. Evaluation of microbiome in primary and permanent dentition in grade C periodontitis in young individuals. J. Periodontol. 2024. [Google Scholar] [CrossRef]
- Shenker, B.J.; Walker, L.P.; Zekavat, A.; Korostoff, J.; Boesze-Battaglia, K. Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin-Induces Cell Cycle Arrest in a Glycogen Synthase Kinase (GSK)-3-Dependent Manner in Oral Keratinocytes. Int. J. Mol. Sci. 2022, 23, 1831. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.F.; Altabtbaei, K.; Kumar, P.S.; Casati, M.Z.; Ruiz, K.G.S.; Sallum, E.A.; Nociti-Junior, F.H.; Casarin, R.C.V. Parents with periodontitis impact the subgingival colonization of their offspring. Sci. Rep. 2021, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.J.; Stamatelakys, C.; Adair, S.M. Resolution of early lesions of juvenile periodontitis with tetracycline therapy alone: Long-term observations of 4 cases. J. Periodontol. 1991, 62, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Mandell, R.L.; Tripodi, L.S.; Savitt, E.; Goodson, J.M.; Socransky, S.S. The effect of treatment on Actinobacillus actinomycetemcomitans in localized juvenile periodontitis. J. Periodontol. 1986, 57, 94–99. [Google Scholar] [CrossRef]
- Miller, K.A.; Branco-de-Almeida, L.S.; Wolf, S.; Hovencamp, N.; Treloar, T.; Harrison, P.; Aukhil, I.; Gong, Y.; Shaddox, L.M. Long-term clinical response to treatment and maintenance of localized aggressive periodontitis: A cohort study. J. Clin. Periodontol. 2017, 44, 158–168. [Google Scholar] [CrossRef]
- Schreiner, H.C.; Sinatra, K.; Kaplan, J.B.; Furgang, D.; Kachlany, S.C.; Planet, P.J.; Perez, B.A.; Figurski, D.H.; Fine, D.H. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc. Natl. Acad. Sci. USA 2003, 100, 7295–7300. [Google Scholar] [CrossRef]
- Schreiner, H.; Li, Y.; Cline, J.; Tsiagbe, V.K.; Fine, D.H. A comparison of Aggregatibacter actinomycetemcomitans (Aa) virulence traits in a rat model for periodontal disease. PLoS ONE 2013, 8, e69382. [Google Scholar] [CrossRef]
- Parthiban, C.; Varudharasu, D.; Shanmugam, M.; Gopal, P.; Ragunath, C.; Thomas, L.; Nitz, M.; Ramasubbu, N. Structural and functional analysis of de-N-acetylase PgaB from periodontopathogen Aggregatibacter actinomycetemcomitans. Mol. Oral Microbiol. 2017, 32, 324–340. [Google Scholar] [CrossRef]
- Ristow, L.C.; Welch, R.A. RTX Toxins Ambush Immunity’s First Cellular Responders. Toxins 2019, 11, 720. [Google Scholar] [CrossRef]
- Azzi-Martin, L.; Touffait-Calvez, V.; Everaert, M.; Jia, R.; Sifre, E.; Seeneevassen, L.; Varon, C.; Dubus, P.; Menard, A. Cytolethal Distending Toxin Modulates Cell Differentiation and Elicits Epithelial to Mesenchymal Transition. J. Infect. Dis. 2024, 229, 1688–1701. [Google Scholar] [CrossRef]
- Ivanyi, L.; Challacombe, S.J.; Lehner, T. The specificity of serum factors in lymphocyte transformation in periodontal disease. Clin. Exp. Immunol. 1973, 14, 491–500. [Google Scholar] [PubMed]
- Ebersole, J.L.; Dawson, D., 3rd; Emecen-Huja, P.; Nagarajan, R.; Howard, K.; Grady, M.E.; Thompson, K.; Peyyala, R.; Al-Attar, A.; Lethbridge, K.; et al. The periodontal war: Microbes and immunity. Periodontol. 2000 2017, 75, 52–115. [Google Scholar] [CrossRef]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune response: The key to bone resorption in periodontal disease. J. Periodontol. 2005, 76, 2033–2041. [Google Scholar] [CrossRef]
- Genco, R.J.; Sanz, M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol. 2000 2020, 83, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Dean, R.T.; Sanderson, C.J. Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect. Immun. 1980, 28, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Wilton, J.M. Leukotoxicity of an extract from Actinobacillus actinomycetemcomitans for human gingival polymorphonuclear leukocytes. Inflammation 1981, 5, 1–12. [Google Scholar] [CrossRef]
- Proctor, D.M.; Shelef, K.M.; Gonzalez, A.; Davis, C.L.; Dethlefsen, L.; Burns, A.R.; Loomer, P.M.; Armitage, G.C.; Ryder, M.I.; Millman, M.E.; et al. Microbial biogeography and ecology of the mouth and implications for periodontal diseases. Periodontol. 2000 2020, 82, 26–41. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef]
- Tabak, L.A.; Levine, M.J.; Mandel, I.D.; Ellison, S.A. Role of salivary mucins in the protection of the oral cavity. J. Oral Pathol. 1982, 11, 1–17. [Google Scholar] [CrossRef]
- Mandel, I.D. The role of saliva in maintaining oral homeostasis. J. Am. Dent. Assoc. 1989, 119, 298–304. [Google Scholar] [CrossRef]
- Socransky, S.S.; Manganiello, S.D. The oral microbiota of man from birth to senility. J. Periodontol. 1971, 42, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.M.; Yansouni, C.P.; Behr, M.A. Causality and gastrointestinal infections: Koch, Hill, and Crohn’s. Lancet Infect. Dis. 2008, 8, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Ritz, H.L. Microbial population shifts in developing human dental plaque. Arch. Oral Biol. 1967, 12, 1561–1568. [Google Scholar] [CrossRef]
- Hujoel, P.P.; White, B.A.; Garcia, R.I.; Listgarten, M.A. The dentogingival epithelial surface area revisited. J. Periodontal Res. 2001, 36, 48–55. [Google Scholar] [CrossRef]
- Blaser, M.J. Missing Microbes: How the Overuse of Antibiotics is Fueling our Modern Plagues, 1st ed.; Henry Holt and Company: New York, NY, USA, 2014. [Google Scholar]
- Del Romero, J.; Moreno Guillen, S.; Rodriguez-Artalejo, F.J.; Ruiz-Galiana, J.; Canton, R.; De Lucas Ramos, P.; Garcia-Botella, A.; Garcia-Lledo, A.; Hernandez-Sampelayo, T.; Gomez-Pavon, J.; et al. Sexually transmitted infections in Spain: Current status. Rev. Esp. Quimioter. 2023, 36, 444–465. [Google Scholar] [CrossRef] [PubMed]
- Okell, C.C.; Elliott, S.D. Bacteriaaemia and oral sepsis with special reference to the aetiology of subacute endocarditis. Lancet 1935, 869–872. [Google Scholar] [CrossRef]
- Silver, J.G.; Martin, A.W.; McBride, B.C. Experimental transient bacteraemias in human subjects with varying degrees of plaque accumulation and gingival inflammation. J. Clin. Periodontol. 1977, 4, 92–99. [Google Scholar] [CrossRef]
- Wells, P.M.; Sprockett, D.D.; Bowyer, R.C.E.; Kurushima, Y.; Relman, D.A.; Williams, F.M.K.; Steves, C.J. Influential factors of saliva microbiota composition. Sci. Rep. 2022, 12, 18894. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Tsai, C.C.; Baehni, P.C.; Stoller, N.; McArthur, W.P. Interaction of inflammatory cells and oral microorganisms. IV. In vitro release of lysosomal constituents from polymorphonuclear leukocytes exposed to supragingival and subgingival bacterial plaque. Infect. Immun. 1977, 16, 1013–1023. [Google Scholar] [CrossRef]
- Van Dyke, T.E.; Horoszewicz, H.U.; Genco, R.J. The polymorphonuclear leukocyte (PMNL) locomotor defect in juvenile periodontitis. Study of random migration, chemokinesis and chemotaxis. J. Periodontol. 1982, 53, 682–687. [Google Scholar] [CrossRef]
- Fives-Taylor, P.M.; Meyer, D.H.; Mintz, K.P.; Brissette, C. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol. 2000 1999, 20, 136–167. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Schreiner, H. Oral microbial interactions from an ecological perspective: A narrative review. Front. Oral Health 2023, 4, 1229118. [Google Scholar] [CrossRef] [PubMed]
- Aruni, W.; Chioma, O.; Fletcher, H.M. Filifactor alocis: The Newly Discovered Kid on the Block with Special Talents. J. Dent. Res. 2014, 93, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Patil, A.G.; Velusamy, S.K. Aggregatibacter actinomycetemcomitans (Aa) under the Radar: Myths and Misunderstandings of Aa and Its Role in Aggressive Periodontitis. Front. Immunol. 2019, 10, 728. [Google Scholar] [CrossRef]
- Offenbacher, S.; Katz, V.; Fertik, G.; Collins, J.; Boyd, D.; Maynor, G.; McKaig, R.; Beck, J. Periodontal infection as a possible risk factor for preterm low birth weight. J. Periodontol. 1996, 67, 1103–1113. [Google Scholar] [CrossRef]
- Fine, D.H.; Furgang, D.; McKiernan, M.; Tereski-Bischio, D.; Ricci-Nittel, D.; Zhang, P.; Araujo, M.W. An investigation of the effect of an essential oil mouthrinse on induced bacteraemia: A pilot study. J. Clin. Periodontol. 2010, 37, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Riggio, M.P.; Walker, K.F.; MacKenzie, D.; Shearer, B. Bacteraemia following periodontal procedures. J. Clin. Periodontol. 2005, 32, 708–713. [Google Scholar] [CrossRef]
- Shanson, D. New British and American guidelines for the antibiotic prophylaxis of infective endocarditis: Do the changes make sense? A critical review. Curr. Opin. Infect. Dis. 2008, 21, 191–199. [Google Scholar] [CrossRef]
- Bergenholtz, G. Effect of bacterial products on inflammatory reactions in the dental pulp. Scand. J. Dent. Res. 1977, 85, 122–129. [Google Scholar]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. USA 2006, 103, 732–737. [Google Scholar] [CrossRef]
- Black, S.; Phillips, D.; Hickey, J.W.; Kennedy-Darling, J.; Venkataraaman, V.G.; Samusik, N.; Goltsev, Y.; Schurch, C.M.; Nolan, G.P. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc. 2021, 16, 3802–3835. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, J.L.; Dewhirst, F.E.; Borisy, G.G. Biogeographty of the Oral Microbiome: The Site-Specific Hyothesis. Ann. Rev. Microbiol. 2019, 73, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.; Glogauer, M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp. Cell Res. 2016, 342, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Oshrain, H.I.; Salind, A.; Mandel, I.D. A method for collection of subgingival plaque and calculus. J. Periodontol. 1968, 39, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Greene, L.S. Microscopic evaluation of root surface associations in vivo. J. Periodontal Res. 1984, 19, 152–167. [Google Scholar] [CrossRef]
- Wecke, J.; Wolf, V.; Fath, S.; Bernimoulin, J.P. The occurrence of treponemes and their spherical bodies on polytetrafluoroethylene membranes. Oral Microbiol. Immunol. 1995, 10, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Markowitz, K.; Furgang, D.; Velliyagounder, K. Aggregatibacter actinomycetemcomitans as an early colonizer of oral tissues: Epithelium as a reservoir? J. Clin. Microbiol. 2010, 48, 4464–4473. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J.; Koo, H. Oral polymicrobial communities: Assembly, function, and impact on diseases. Cell Host Microbe 2023, 31, 528–538. [Google Scholar] [CrossRef]
- Hajishengallis, G. Interconnection of periodontal disease and comorbidities: Evidence, mechanisms, and implications. Periodontol. 2000 2022, 89, 9–18. [Google Scholar] [CrossRef]
- Haubek, D.; Poulsen, K.; Kilian, M. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect. Immun. 2007, 75, 3080–3088. [Google Scholar] [CrossRef]
- Fine, D.H.; Toruner, G.A.; Velliyagounder, K.; Sampathkumar, V.; Godboley, D.; Furgang, D. A lactotransferrin single nucleotide polymorphism demonstrates biological activity that can reduce susceptibility to caries. Infect. Immun. 2013, 81, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilyfard, R.; Bonyadifard, H.; Paknahad, M. Dental Caries Detection and Classification in CBCT Images Using Deep Learning. Int. Dent. J. 2024, 74, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Pitts, N.B.; Twetman, S.; Fisher, J.; Marsh, P.D. Understanding dental caries as a non-communicable disease. Br. Dent. J. 2021, 231, 749–753. [Google Scholar] [CrossRef]
- Sioson, P.B.; Furgang, D.; Steinberg, L.M.; Fine, D.H. Proximal caries in juvenile periodontitis patients. J. Periodontol. 2000, 71, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Zambon, J.J. Actinobacillus actinomycetemcomitans in human periodontal disease. J. Clin. Periodontol. 1985, 12, 1–20. [Google Scholar] [CrossRef]
- Holtfreter, B.; Kuhr, K.; Borof, K.; Tonetti, M.S.; Sanz, M.; Kornman, K.; Jepsen, S.; Aarabi, G.; Volzke, H.; Kocher, T.; et al. ACES: A new framework for the application of the 2018 periodontal status classification scheme to epidemiological survey data. J. Clin. Periodontol. 2024, 51, 512–521. [Google Scholar] [CrossRef]
- Herzberg, M.C. Platelet-streptococcal interactions in endocarditis. Crit. Rev. Oral Biol. Med. 1996, 7, 222–236. [Google Scholar] [CrossRef]
- Peters, J.; Robinson, F.; Dasco, C.; Gentry, L.O. Subacute bacterial endocarditis due to Actinobacillus actinomycetemcomitans. Am. J. Med. Sci. 1983, 286, 35–41. [Google Scholar] [CrossRef]
- Diamond, I.R.; Grant, R.C.; Feldman, B.M.; Pencharz, P.B.; Ling, S.C.; Moore, A.M.; Wales, P.W. Defining consensus: A systematic review recommends methodologic criteria for reporting of Delphi studies. J. Clin. Epidemiol. 2014, 67, 401–409. [Google Scholar] [CrossRef]
- Hutchinson, P.J.; Kolias, A.G.; Tajsic, T.; Adeleye, A.; Aklilu, A.T.; Apriawan, T.; Bajamal, A.H.; Barthelemy, E.J.; Devi, B.I.; Bhat, D.; et al. Consensus statement from the International Consensus Meeting on the Role of Decompressive Craniectomy in the Management of Traumatic Brain Injury: Consensus statement. Acta Neurochir. 2019, 161, 1261–1274. [Google Scholar] [CrossRef]
- Pelosi, L.; Aranyi, Z.; Beekman, R.; Bland, J.; Coraci, D.; Hobson-Webb, L.D.; Padua, L.; Podnar, S.; Simon, N.; van Alfen, N.; et al. Expert consensus on the combined investigation of ulnar neuropathy at the elbow using electrodiagnostic tests and nerve ultrasound. Clin. Neurophysiol. 2021, 132, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Clyne, B.; Sharp, M.K.; O’Neill, M.; Pollock, D.; Lynch, R.; Amog, K.; Ryan, M.; Smith, S.M.; Mahtani, K.; Booth, A.; et al. An international modified Delphi process supported updating the web-based “right review” tool. J. Clin. Epidemiol. 2024, 170, 111333. [Google Scholar] [CrossRef] [PubMed]
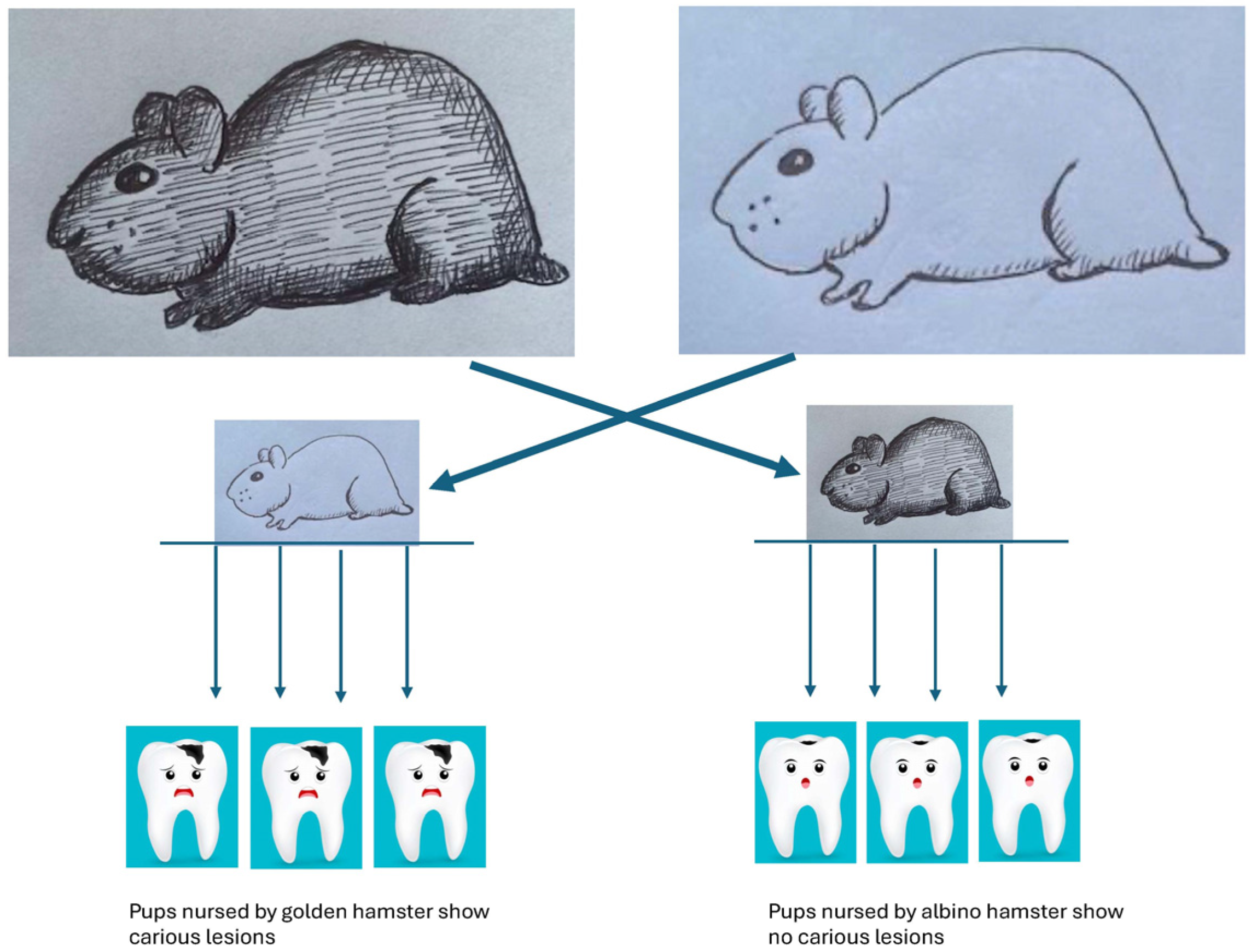

| Disease Category | Pub Med Years | Web of Science Years | Scopus Years | |||
|---|---|---|---|---|---|---|
| 2012–2017 | 2018–2023 | 2012–2017 | 2018–2023 | 2012–2017 | 2018–2023 | |
| Periodontitis | 6492 | 6287 | 10,181 | 16,420 | 13,000 | 18,926 |
| Aggressive Periodontitis | 884 | 534 | 1053 | 817 | 1030 | 679 |
| Localized Aggressive Periodontitis | 179 | 114 | 134 | 102 | 118 | 80 |
| * Stage III Grade C Periodontitis | * 148 | * 138 | * 135 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fine, D.H.; Schreiner, H.; Diehl, S.R. A Rose by Any Other Name: The Long Intricate History of Localized Aggressive Periodontitis. Pathogens 2024, 13, 849. https://doi.org/10.3390/pathogens13100849
Fine DH, Schreiner H, Diehl SR. A Rose by Any Other Name: The Long Intricate History of Localized Aggressive Periodontitis. Pathogens. 2024; 13(10):849. https://doi.org/10.3390/pathogens13100849
Chicago/Turabian StyleFine, Daniel H., Helen Schreiner, and Scott R. Diehl. 2024. "A Rose by Any Other Name: The Long Intricate History of Localized Aggressive Periodontitis" Pathogens 13, no. 10: 849. https://doi.org/10.3390/pathogens13100849
APA StyleFine, D. H., Schreiner, H., & Diehl, S. R. (2024). A Rose by Any Other Name: The Long Intricate History of Localized Aggressive Periodontitis. Pathogens, 13(10), 849. https://doi.org/10.3390/pathogens13100849