

Regulation of R-Loops in DNA Tumor Viruses

{kind=link}
{kind=link}
Abstract
:1. Introduction
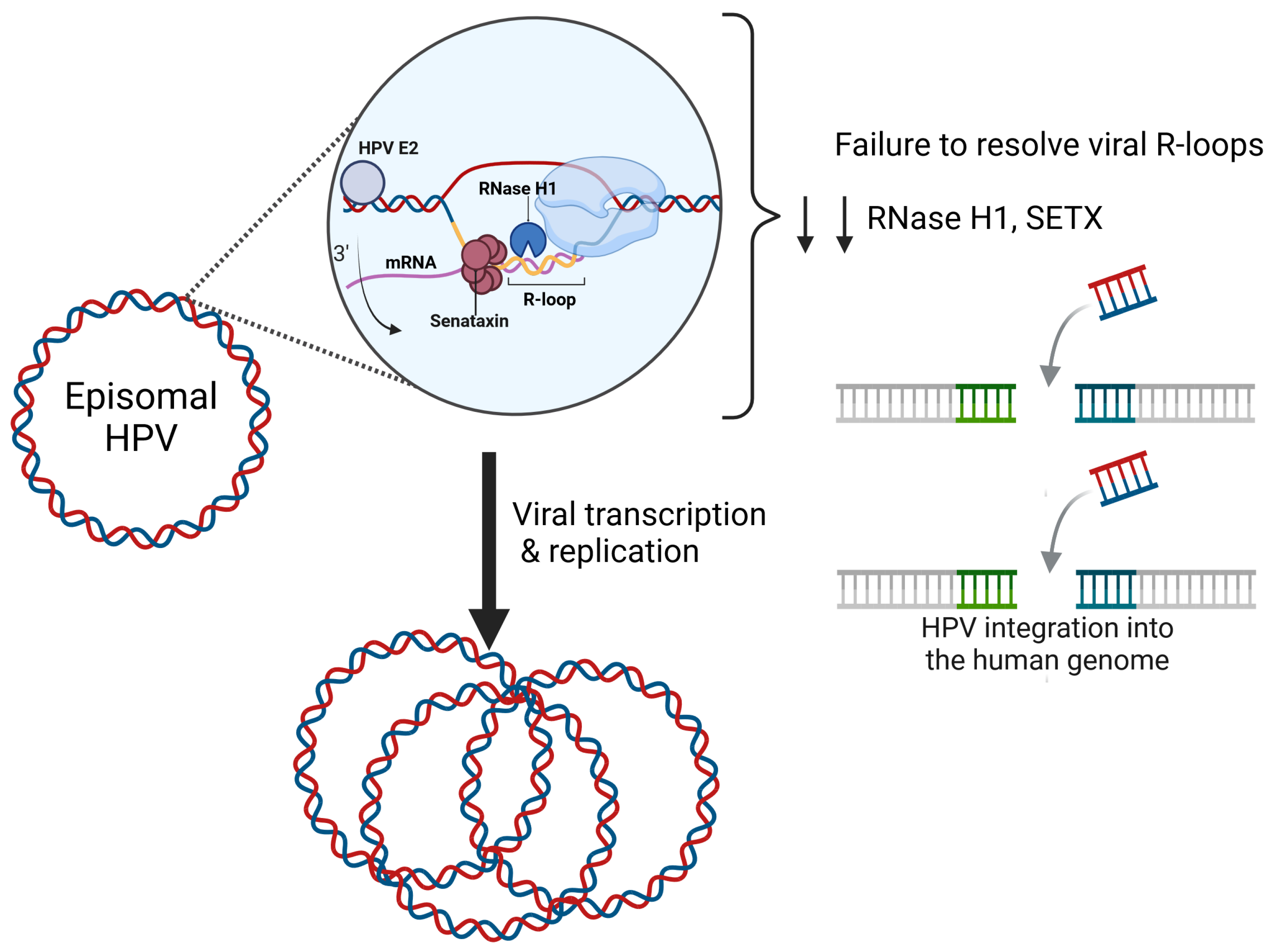
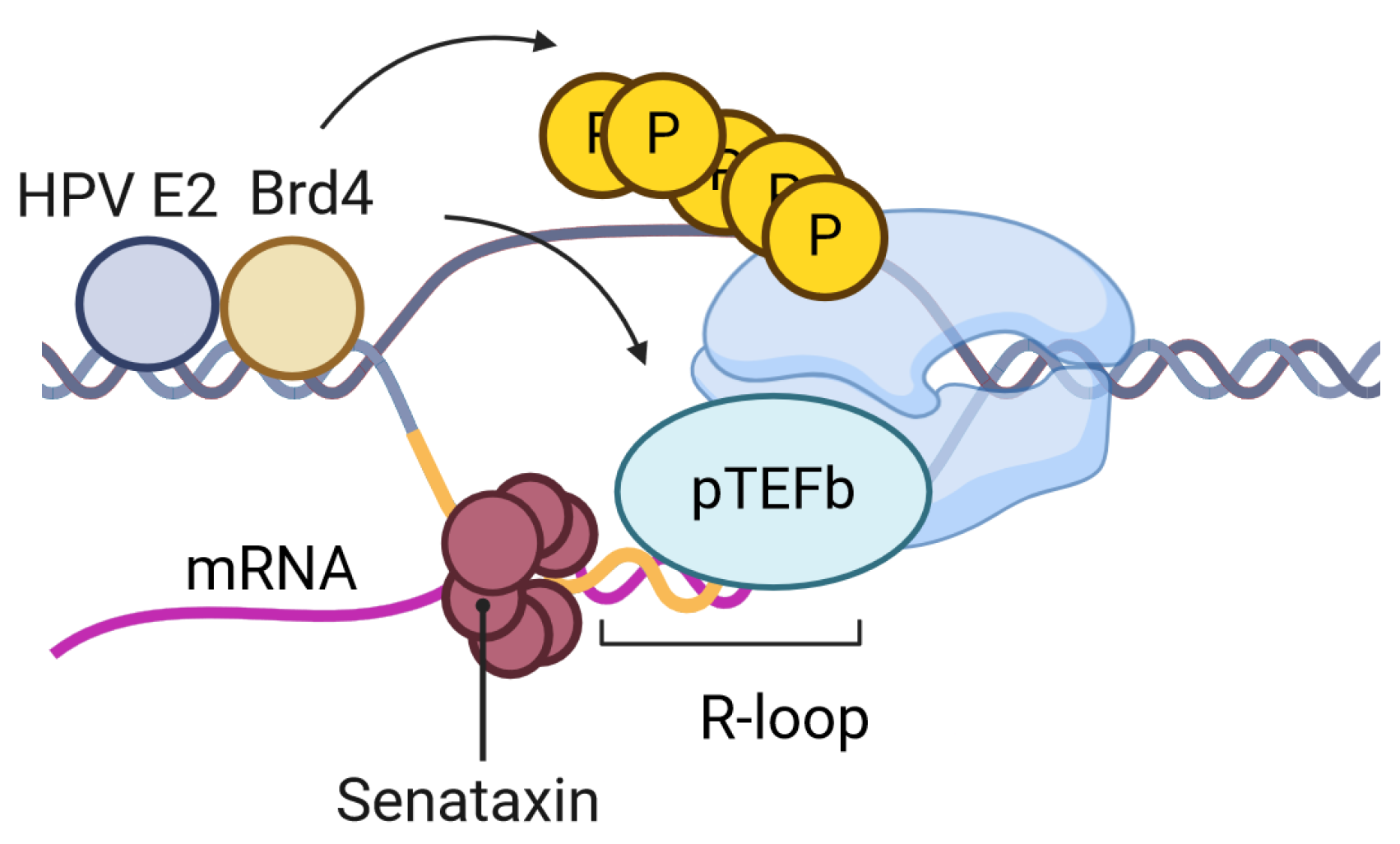
2. Human Papillomavirus (HPV)
3. Epstein–Barr Virus (EBV)
4. Kaposi Sarcoma-Associated Herpesvirus (KSHV)
5. Hepatitis B Virus (HBV)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Allison, D.F.; Wang, G.G. R-loops: Formation, function, and relevance to cell stress. Cell Stress 2019, 3, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Elsakrmy, N.; Cui, H. R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 7064. [Google Scholar] [CrossRef] [PubMed]
- Malig, M.; Hartono, S.R.; Giafaglione, J.M.; Sanz, L.A.; Chedin, F. Ultra-deep Coverage Single-molecule R-loop Footprinting Reveals Principles of R-loop Formation. J. Mol. Biol. 2020, 432, 2271–2288. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.J.; Hamperl, S.; Swigut, T.; Cimprich, K.A. qDRIP: A method to quantitatively assess RNA-DNA hybrid formation genome-wide. Nucleic Acids Res. 2020, 48, e84. [Google Scholar] [CrossRef]
- Xu, Y.; Jiao, Y.; Liu, C.; Miao, R.; Liu, C.; Wang, Y.; Ma, C.; Liu, J. R-loop and diseases: The cell cycle matters. Mol. Cancer 2024, 23, 84. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Mischo, H.E.; Gómez-González, B.; Grzechnik, P.; Rondón, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin Associates with Replication Forks to Protect Fork Integrity across RNA-Polymerase-II-Transcribed Genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Achar, Y.J.; Piccini, D.; Galanti, L.; Colosio, A.; Foiani, M.; Liberi, G. Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 2018, 46, 1227–1239. [Google Scholar] [CrossRef]
- Gatti, V.; De Domenico, S.; Melino, G.; Peschiaroli, A. Senataxin and R-loops homeostasis: Multifaced implications in carcinogenesis. Cell Death Discov. 2023, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, A.; Pires, V.B.; Bento, F.; Kellner, V.; Luke-Glaser, S.; Yakoub, G.; Ulrich, H.D.; Luke, B. RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cell Rep. 2019, 29, 2890–2900.e2895. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Sakhuja, K.; Crouch, R.J. RNase H1, the Gold Standard for R-Loop Detection. Methods Mol. Biol. 2022, 2528, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, Y.A.; Fernando, C.M.; Tran, E.J. The balancing act of R-loop biology: The good, the bad, and the ugly. J. Biol. Chem. 2020, 295, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Chédin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA–RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019, 14, 1734–1755. [Google Scholar] [CrossRef]
- Miller, H.E.; Montemayor, D.; Li, J.; Levy, S.A.; Pawar, R.; Hartono, S.; Sharma, K.; Frost, B.; Chedin, F.; Bishop, A.J.R. Exploration and analysis of R-loop mapping data with RLBase. Nucleic Acids Res. 2022, 51, D1129–D1137. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef]
- Thomas, M.; Pim, D.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Templeton, C.W.; Laimins, L.A. p53-dependent R-loop formation and HPV pathogenesis. Proc. Natl. Acad. Sci. USA 2023, 120, e2305907120. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Smith, K.; Crowner, A.; Androphy, E.J.; DeSmet, M. Senataxin mediates R-loop resolution on HPV episomes. J. Virol. 2024, 98, e0100324. [Google Scholar] [CrossRef] [PubMed]
- Templeton, C.W.; Laimins, L.A. HPV induced R-loop formation represses innate immune gene expression while activating DNA damage repair pathways. PLoS Pathog. 2024, 20, e1012454. [Google Scholar] [CrossRef] [PubMed]
- Panatta, E.; Butera, A.; Mammarella, E.; Pitolli, C.; Mauriello, A.; Leist, M.; Knight, R.A.; Melino, G.; Amelio, I. Metabolic regulation by p53 prevents R-loop-associated genomic instability. Cell Rep. 2022, 41, 111568. [Google Scholar] [CrossRef]
- Chow, L.T.; Reilly, S.S.; Broker, T.R.; Taichman, L.B. Identification and mapping of human papillomavirus type 1 RNA transcripts recovered from plantar warts and infected epithelial cell cultures. J. Virol. 1987, 61, 1913–1918. [Google Scholar] [CrossRef]
- Dooley, K.E.; Warburton, A.; McBride, A.A. Tandemly Integrated HPV16 Can Form a Brd4-Dependent Super-Enhancer-Like Element That Drives Transcription of Viral Oncogenes. mBio 2016, 7, 10-1128. [Google Scholar] [CrossRef]
- Thomas, Y.; Androphy, E.J. Acetylation of E2 by P300 Mediates Topoisomerase Entry at the Papillomavirus Replicon. J. Virol. 2019, 93, e02224-18. [Google Scholar] [CrossRef]
- Hu, Y.; Clower, R.V.; Melendy, T. Cellular topoisomerase I modulates origin binding by bovine papillomavirus type 1 E1. J. Virol. 2006, 80, 4363–4371. [Google Scholar] [CrossRef]
- Sakasai, R.; Isono, M.; Wakasugi, M.; Hashimoto, M.; Sunatani, Y.; Matsui, T.; Shibata, A.; Matsunaga, T.; Iwabuchi, K. Aquarius is required for proper CtIP expression and homologous recombination repair. Sci. Rep. 2017, 7, 13808. [Google Scholar] [CrossRef]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef] [PubMed]
- Hubert, W.G.; Kanaya, T.; Laimins, L.A. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5′ of the minimal origin. J. Virol. 1999, 73, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential Requirements for Conserved E2 Binding Sites in the Life Cycle of Oncogenic Human Papillomavirus Type 31. J. Virol. 1998, 72, 1071. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Kannan, A.; Jiang, X.; He, L.; Ahmad, S.; Gangwani, L. ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain 2020, 143, 69–93. [Google Scholar] [CrossRef]
- Kannan, A.; Gangadharan Leela, S.; Branzei, D.; Gangwani, L. Role of senataxin in R-loop-mediated neurodegeneration. Brain Commun. 2024, 6, fcae239. [Google Scholar] [CrossRef]
- Kannan, A.; Cuartas, J.; Gangwani, P.; Branzei, D.; Gangwani, L. Mutation in senataxin alters the mechanism of R-loop resolution in amyotrophic lateral sclerosis 4. Brain 2022, 145, 3072–3094. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous Recombination Repair Factors Rad51 and BRCA1 Are Necessary for Productive Replication of Human Papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A.; Khurana, S. Multiple Roles of Brd4 in the Infectious Cycle of Human Papillomaviruses. Front. Mol. Biosci. 2021, 8, 725794. [Google Scholar] [CrossRef] [PubMed]
- Iftner, T.; Haedicke-Jarboui, J.; Wu, S.Y.; Chiang, C.M. Involvement of Brd4 in different steps of the papillomavirus life cycle. Virus Res. 2017, 231, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Androphy, E.J.; DeSmet, M. SETD6 Regulates E2-Dependent Human Papillomavirus Transcription. J. Virol. 2022, 96, e0129522. [Google Scholar] [CrossRef]
- Helfer, C.M.; Yan, J.; You, J. The cellular bromodomain protein Brd4 has multiple functions in E2-mediated papillomavirus transcription activation. Viruses 2014, 6, 3228–3249. [Google Scholar] [CrossRef] [PubMed]
- DeSmet, M.; Jose, L.; Isaq, N.; Androphy, E.J. Phosphorylation of a Conserved Tyrosine in the Papillomavirus E2 Protein Regulates Brd4 Binding and Viral Replication. J. Virol. 2019, 93, e01801-18. [Google Scholar] [CrossRef]
- To, K.K.W.; Xing, E.; Larue, R.C.; Li, P.K. BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications. Molecules 2023, 28, 3043. [Google Scholar] [CrossRef]
- Sarott, R.C.; You, I.; Li, Y.D.; Toenjes, S.T.; Donovan, K.A.; Seo, P.; Ordonez, M.; Byun, W.S.; Hassan, M.M.; Wachter, F.; et al. Chemical Specification of E3 Ubiquitin Ligase Engagement by Cysteine-Reactive Chemistry. J. Am. Chem. Soc. 2023, 145, 21937–21944. [Google Scholar] [CrossRef]
- Hu, J.; Hu, B.; Xu, F.; Wang, M.; Qin, C.; McEachern, D.; Stuckey, J.; Wang, S. Precise Conformational Control Yielding Highly Potent and Exceptionally Selective BRD4 Degraders with Strong Antitumor Activity. J. Med. Chem. 2023, 66, 8222–8237. [Google Scholar] [CrossRef]
- Lam, F.C.; Kong, Y.W.; Huang, Q.; Vu Han, T.L.; Maffa, A.D.; Kasper, E.M.; Yaffe, M.B. BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat. Commun. 2020, 11, 4083. [Google Scholar] [CrossRef]
- Edwards, D.S.; Maganti, R.; Tanksley, J.P.; Luo, J.; Park, J.J.H.; Balkanska-Sinclair, E.; Ling, J.; Floyd, S.R. BRD4 Prevents R-Loop Formation and Transcription-Replication Conflicts by Ensuring Efficient Transcription Elongation. Cell Rep. 2020, 32, 108166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Gold, S.; Iwanaszko, M.; Howard, B.C.; Wang, L.; Shilatifard, A. Distinct layers of BRD4-PTEFb reveal bromodomain-independent function in transcriptional regulation. Mol. Cell 2023, 83, 2896–2910.e2894. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757.e745. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed]
- Itzen, F.; Greifenberg, A.K.; Bösken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef]
- Rennekamp, A.J.; Lieberman, P.M. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. J. Virol. 2011, 85, 2837–2850. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yajima, M.; Ikuta, K. Epstein-Barr virus strain variation and cancer. Cancer Sci. 2019, 110, 1132–1139. [Google Scholar] [CrossRef]
- Hammerschmidt, W.; Sugden, B. Replication of Epstein-Barr viral DNA. Cold Spring Harb. Perspect. Biol. 2013, 5, a013029. [Google Scholar] [CrossRef]
- Yetming, K.D.; Lupey-Green, L.N.; Biryukov, S.; Hughes, D.J.; Marendy, E.M.; Miranda, J.L.; Sample, J.T. The BHLF1 Locus of Epstein-Barr Virus Contributes to Viral Latency and B-Cell Immortalization. J. Virol. 2020, 94, e01215-20. [Google Scholar] [CrossRef]
- Peng, X.P.; Zhao, X. The multi-functional Smc5/6 complex in genome protection and disease. Nat. Struct. Mol. Biol. 2023, 30, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Lafuente-Barquero, J.; Luke-Glaser, S.; Graf, M.; Silva, S.; Gómez-González, B.; Lockhart, A.; Lisby, M.; Aguilera, A.; Luke, B. The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLoS Genet. 2017, 13, e1007136. [Google Scholar] [CrossRef] [PubMed]
- Bentley, P.; Tan, M.J.A.; McBride, A.A.; White, E.A.; Howley, P.M. The SMC5/6 Complex Interacts with the Papillomavirus E2 Protein and Influences Maintenance of Viral Episomal DNA. J. Virol. 2018, 92, e00356-18. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and Characterizing Interplay between Hepatitis B Virus X Protein and Smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef]
- Gibson, R.T.; Androphy, E.J. The SMC5/6 Complex Represses the Replicative Program of High-Risk Human Papillomavirus Type 31. Pathogens 2020, 9, 786. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Funato, K.; Miyakawa, Y.; Tanaka, E.; Seimiya, T.; Yamagami, M.; Tsutsumi, T.; Okushin, K.; Miyakawa, K.; et al. HBx-induced degradation of Smc5/6 complex impairs homologous recombination-mediated repair of damaged DNA. J. Hepatol. 2022, 76, 53–62. [Google Scholar] [CrossRef]
- Yiu, S.P.T.; Guo, R.; Zerbe, C.; Weekes, M.P.; Gewurz, B.E. Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep. 2022, 38, 110411. [Google Scholar] [CrossRef]
- Juillard, F.; Tan, M.; Li, S.; Kaye, K.M. Kaposi’s Sarcoma Herpesvirus Genome Persistence. Front. Microbiol. 2016, 7, 1149. [Google Scholar] [CrossRef]
- Grundhoff, A.; Ganem, D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 2003, 77, 2779–2783. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of Kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhou, F.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 2004, 64, 4064–4068. [Google Scholar] [CrossRef]
- Jackson, B.R.; Noerenberg, M.; Whitehouse, A. A novel mechanism inducing genome instability in Kaposi’s sarcoma-associated herpesvirus infected cells. PLoS Pathog. 2014, 10, e1004098. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Zheng, Z.M. KSHV ORF57, a protein of many faces. Viruses 2015, 7, 604–633. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Colgan, K.J.; Whitehouse, A. Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog. 2008, 4, e1000194. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Gupta, A.; Jenjaroenpun, P.; Owens, S.; Forrest, J.C.; Nookaew, I. R-loop-forming Sequences Analysis in Thousands of Viral Genomes Identify A New Common Element in Herpesviruses. Sci. Rep. 2020, 10, 6389. [Google Scholar] [CrossRef]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef]
- Gómez-Moreno, A.; Garaigorta, U. Hepatitis B Virus and DNA Damage Response: Interactions and Consequences for the Infection. Viruses 2017, 9, 304. [Google Scholar] [CrossRef]
- Broennimann, K.; Ricardo-Lax, I.; Adler, J.; Shaul, Y. Evidence for a Hepatitis B Virus Short RNA Fragment Directly Targeting the Cellular RRM2 Gene. Cells 2022, 11, 2248. [Google Scholar] [CrossRef]
- Matos, D.A.; Zhang, J.M.; Ouyang, J.; Nguyen, H.D.; Genois, M.M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2020, 77, 514–527.e514. [Google Scholar] [CrossRef]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martinez, S.; Tsanov, N.; Abou Merhi, R.; Maiorano, D. An ATR‐dependent function for the Ddx19 RNA helicase in nuclear R‐loop metabolism. EMBO J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV when Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Vats, A.; Chimankar, V. MCV Truncated Large T antigen interacts with BRD4 in tumors. Matters 2019, 2019. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Schowalter, R.M.; Jiao, J.; Buck, C.B.; You, J. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 2012, 8, e1003021. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, R.; Sathira, N.P.; Kanai, A.; Tanimoto, K.; Arauchi, T.; Tanaka, Y.; Hashimoto, S.; Sugano, S.; Nakai, K.; Suzuki, Y. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res. 2011, 21, 775–789. [Google Scholar] [CrossRef]
- Mackay, R.P.; Xu, Q.; Weinberger, P.M. R-Loop Physiology and Pathology: A Brief Review. DNA Cell Biol. 2020, 39, 1914–1925. [Google Scholar] [CrossRef]
- Yu, K.; Chedin, F.; Hsieh, C.-L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef]
- Giam, C.Z.; Pasupala, N. NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells. Viruses 2022, 14, 877. [Google Scholar] [CrossRef]
- Rogan, P.K.; Mucaki, E.J.; Shirley, B.C. A proposed molecular mechanism for pathogenesis of severe RNA-viral pulmonary infections. F1000Research 2020, 9, 943. [Google Scholar] [CrossRef]
- Boehmer, P.E. RNA binding and R-loop formation by the herpes simplex virus type-1 single-stranded DNA-binding protein (ICP8). Nucleic Acids Res. 2004, 32, 4576–4584. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, D.; Jeong, J.; Lee, S.; Kim, S.; Ahn, K. Human immunodeficiency virus-1 induces and targets host genomic R-loops for viral genome integration. Microbiol. Infect. Dis. 2024. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crowner, A.; Smith, K.; DeSmet, M. Regulation of R-Loops in DNA Tumor Viruses. Pathogens 2024, 13, 863. https://doi.org/10.3390/pathogens13100863
Crowner A, Smith K, DeSmet M. Regulation of R-Loops in DNA Tumor Viruses. Pathogens. 2024; 13(10):863. https://doi.org/10.3390/pathogens13100863
Chicago/Turabian StyleCrowner, Anaiya, Keely Smith, and Marsha DeSmet. 2024. "Regulation of R-Loops in DNA Tumor Viruses" Pathogens 13, no. 10: 863. https://doi.org/10.3390/pathogens13100863