
Hybrid-Capture Target Enrichment in Human Pathogens: Identification, Evolution, Biosurveillance, and Genomic Epidemiology


{kind=link}
{kind=link}
Abstract
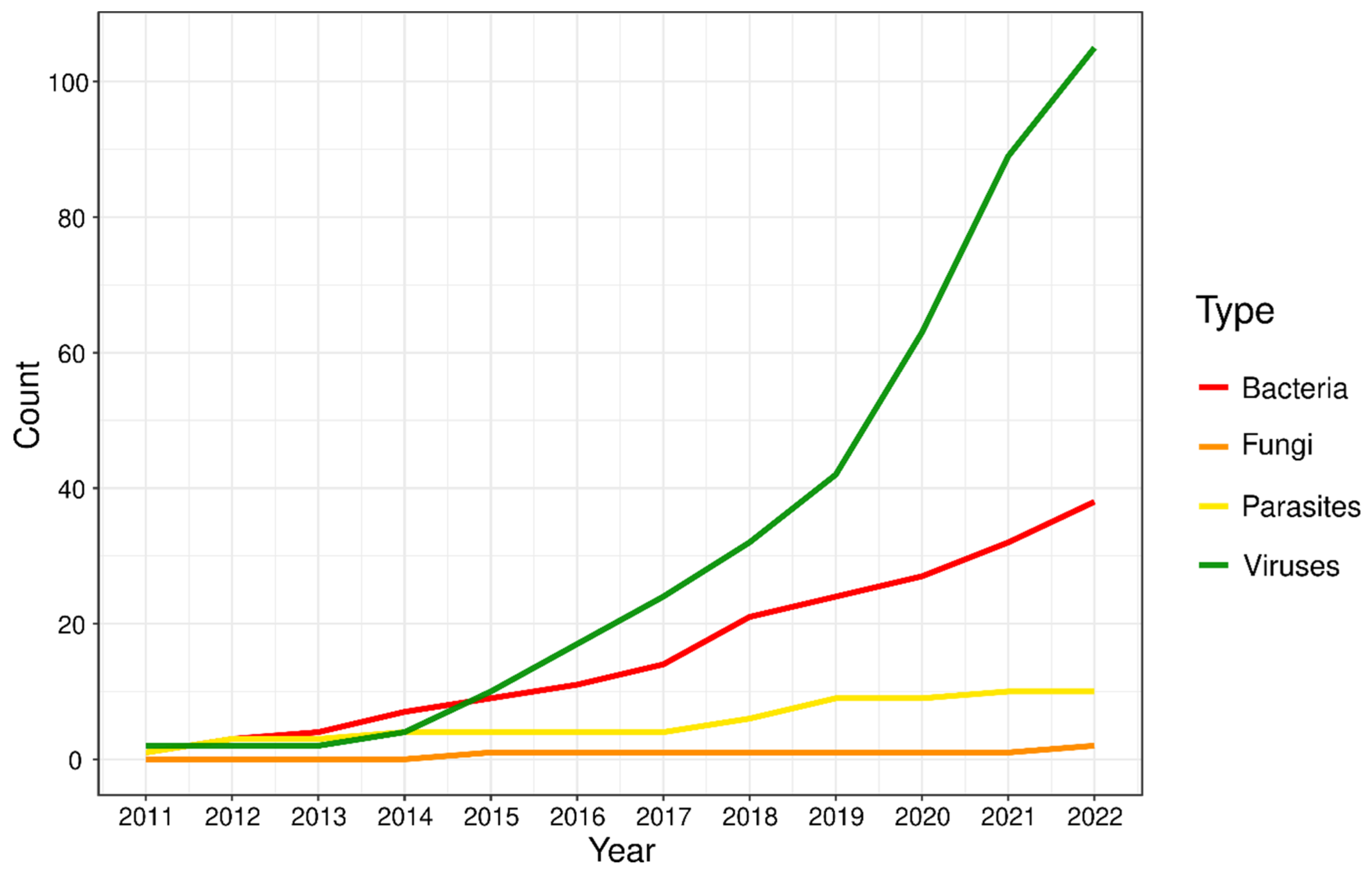
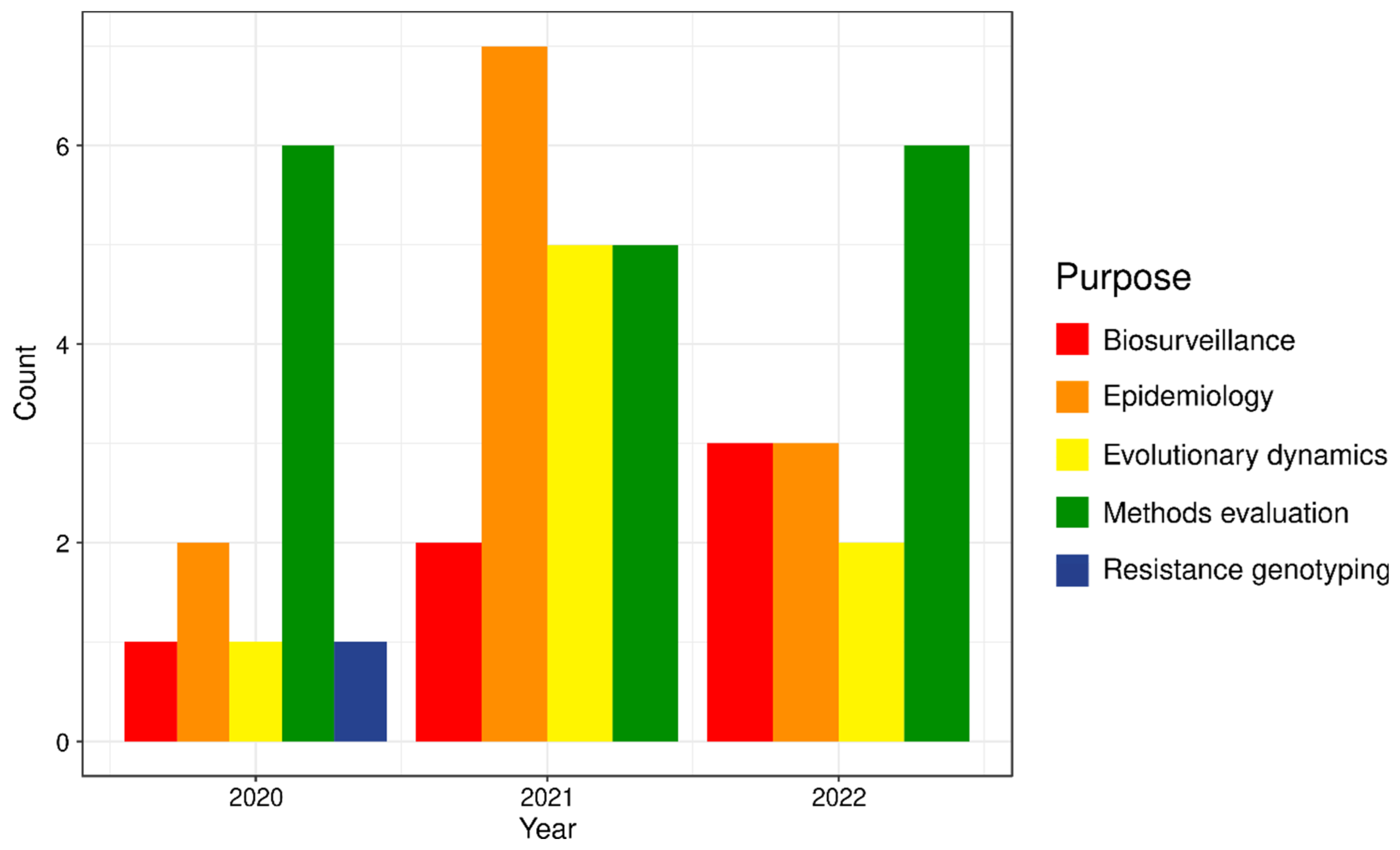
:1. Introduction
2. Hybrid-capture target enrichment for Pathogens
2.1. Bacteria
2.1.1. Hybrid Capture versus Amplicon Sequencing in 16S rRNA
2.1.2. Antimicrobial Resistance
2.1.3. Genome Characterisation of Fastidious Bacteria Using Hybrid Capture
2.1.4. Broad Spectrum Bacteria Bait Panels
2.2. Viruses
2.2.1. Oncoviruses
2.2.2. Human Immunodeficiency Virus
2.2.3. Unravelling Discrepancies in Genomes between Clinical and Cultured Samples in Herpesviruses
2.2.4. Broad-Range Viral Bait Panels and Clinical Metagenomics
2.3. Parasites
2.3.1. Malaria
2.3.2. Neglected Tropical Diseases (NTD)
2.3.3. Hybrid Capture and the Future of Neglected Tropical Diseases
2.4. Fungi
3. Biosurveillance and Emerging Viral Infectious Diseases
3.1. Arthropod-Borne Viruses
3.1.1. Mosquito-Borne Viruses
3.1.2. Tick-Borne Viruses
3.2. Mammalian Zoonotic Viruses
3.2.1. Lassa and Ebola Viruses
3.2.2. Coronaviruses
3.2.3. Monkeypox Virus
3.2.4. Moving Forward: Biosurveillance of Mammals
4. Case Study: SARS-CoV-2
5. The Future of Hybrid Capture
5.1. Bait Design
5.2. Long-Read Sequencing
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabat, A.J.; Budimir, A.; Nashev, D.; Sá-Leão, R.; van Dijl, J.M.; Laurent, F.; Grundmann, H.; Friedrich, A.W.; ESCMID Study Group of Epidemiological Markers (ESGEM). Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 2013, 18, 20380. [Google Scholar] [CrossRef]
- Frey, K.G.; Bishop-Lilly, K.A. Chapter 15—Next-generation sequencing for pathogen detection and identification. In Methods in Microbiology; Sails, A., Tang, Y.-W., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 42, Chapter 15; pp. 525–554. [Google Scholar]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Govender, K.N.; Street, T.L.; Sanderson, N.D.; Eyre, D.W. Metagenomic sequencing as a pathogen-agnostic clinical diagnostic tool for infectious diseases: A systematic review and meta-analysis of diagnostic test accuracy studies. J. Clin. Microbiol. 2021, 59, e0291620. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using genomics to track global antimicrobial resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Q.; Dong, Y.-Q.; Yin, J.; Qiu, Y.-Q. Diagnostic accuracy of metagenomic next-generation sequencing in diagnosing infectious diseases: A meta-analysis. Sci. Rep. 2022, 12, 21032. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Suarez, C.J.; Banaei, N.; Pinsky, B.A. Next-generation sequencing for infectious disease diagnosis and management: A report of the association for molecular pathology. J. Mol. Diagn. 2015, 17, 623–634. [Google Scholar] [CrossRef]
- Hasan, M.R.; Rawat, A.; Tang, P.; Jithesh, P.V.; Thomas, E.; Tan, R.; Tilley, P. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J. Clin. Microbiol. 2016, 54, 919–927. [Google Scholar] [CrossRef]
- Peker, N.; Garcia-Croes, S.; Dijkhuizen, B.; Wiersma, H.H.; van Zanten, E.; Wisselink, G.; Friedrich, A.W.; Kooistra-Smid, M.; Sinha, B.; Rossen, J.W.A.; et al. A comparison of three different bioinformatics analyses of the 16S-23S rRNA encoding region for bacterial identification. Front. Microbiol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Iwen, P.C.; Hinrichs, S.H.; Rupp, M.E. Utilization of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med. Mycol. 2002, 40, 87–109. [Google Scholar] [CrossRef]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted sequencing approach and its clinical applications for the molecular diagnosis of human diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef]
- Harris, H.M.B.; Hill, C. A place for viruses on the tree of life. Front. Microbiol. 2021, 11, 604048. [Google Scholar] [CrossRef] [PubMed]
- Andermann, T.; Torres Jiménez, M.F.; Matos-Maraví, P.; Batista, R.; Blanco-Pastor, J.L.; Gustafsson, A.L.S.; Kistler, L.; Liberal, I.M.; Oxelman, B.; Bacon, C.D.; et al. A guide to carrying out a phylogenomic target sequence capture project. Front. Genet. 2020, 10, 1407. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, M.; Desnues, C. Hybrid capture-based next generation sequencing and its application to human infectious diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, M.A.; Bos, K.I.; Herbig, A.; Krause, J. Ancient pathogen genomics as an emerging tool for infectious disease research. Nat. Rev. Genet. 2019, 20, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Bos, K.I.; Kühnert, D.; Herbig, A.; Esquivel-Gomez, L.R.; Andrades Valtueña, A.; Barquera, R.; Giffin, K.; Kumar Lankapalli, A.; Nelson, E.A.; Sabin, S.; et al. Paleomicrobiology: Diagnosis and evolution of ancient pathogens. Ann. Rev. Microbiol. 2019, 73, 639–666. [Google Scholar] [CrossRef]
- Singh, R.R. Target enrichment approaches for next-generation sequencing applications in oncology. Diagnostics 2022, 12, 1539. [Google Scholar] [CrossRef]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef]
- Andrades Valtueña, A.; Mittnik, A.; Key, F.M.; Haak, W.; Allmäe, R.; Belinskij, A.; Daubaras, M.; Feldman, M.; Jankauskas, R.; Janković, I.; et al. The Stone Age plague and its persistence in Eurasia. Curr. Biol. 2017, 27, 3683–3691.e8. [Google Scholar] [CrossRef]
- Louca, S.; Mazel, F.; Doebeli, M.; Parfrey, L.W. A census-based estimate of Earth’s bacterial and archaeal diversity. PLoS Biol. 2019, 17, e3000106. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Coupland, P.; Browne, H.P.; Lawley, T.D.; Francis, S.C.; Parkhill, J. Evaluation of PacBio sequencing for full-length bacterial 16S rRNA gene classification. BMC Microbiol. 2016, 16, 274. [Google Scholar] [CrossRef] [PubMed]
- Rassoulian Barrett, S.; Hoffman, N.G.; Rosenthal, C.; Bryan, A.; Marshall, D.A.; Lieberman, J.; Greninger, A.L.; Peddu, V.; Cookson, B.T.; Salipante, S.J. Sensitive identification of bacterial DNA in clinical specimens by broad-range 16S rRNA gene enrichment. J. Clin. Microbiol. 2020, 58, e01605-20. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, M.S.; Wang, J.; Kieran, T.J.; Thomas, J.; Bayona-Vásquez, N.J.; Gao, B.; Devault, A.; Brunelle, B.; Lu, K.; Wang, J.-S.; et al. Improved microbial community characterization of 16S rRNA via metagenome hybridization capture enrichment. Front. Microbiol. 2021, 12, 644662. [Google Scholar] [CrossRef]
- Gupta, S.; Mortensen, M.S.; Schjørring, S.; Trivedi, U.; Vestergaard, G.; Stokholm, J.; Bisgaard, H.; Krogfelt, K.A.; Sørensen, S.J. Amplicon sequencing provides more accurate microbiome information in healthy children compared to culturing. Commun. Biol. 2019, 2, 291. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Iskandar, K.; Murugaiyan, J.; Hammoudi Halat, D.; Hage, S.E.; Chibabhai, V.; Adukkadukkam, S.; Roques, C.; Molinier, L.; Salameh, P.; Van Dongen, M. Antibiotic discovery and resistance: The chase and the race. Antibiotics 2022, 11, 182. [Google Scholar] [CrossRef]
- Baker, S.; Thomson, N.; Weill, F.-X.; Holt, K.E. Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens. Science 2018, 360, 733–738. [Google Scholar] [CrossRef]
- Tunstall, T.; Portelli, S.; Phelan, J.; Clark, T.G.; Ascher, D.B.; Furnham, N. Combining structure and genomics to understand antimicrobial resistance. Comput. Struct. Biotechnol. J. 2020, 18, 3377–3394. [Google Scholar] [CrossRef]
- Noyes, N.R.; Weinroth, M.E.; Parker, J.K.; Dean, C.J.; Lakin, S.M.; Raymond, R.A.; Rovira, P.; Doster, E.; Abdo, Z.; Martin, J.N.; et al. Enrichment allows identification of diverse, rare elements in metagenomic resistome-virulome sequencing. Microbiome 2017, 5, 142. [Google Scholar] [CrossRef]
- Lanza, V.F.; Baquero, F.; Martínez, J.L.; Ramos-Ruíz, R.; González-Zorn, B.; Andremont, A.; Sánchez-Valenzuela, A.; Ehrlich, S.D.; Kennedy, S.; Ruppé, E.; et al. In-depth resistome analysis by targeted metagenomics. Microbiome 2018, 6, 11. [Google Scholar] [CrossRef]
- Guitor, A.K.; Raphenya, A.R.; Klunk, J.; Kuch, M.; Alcock, B.; Surette, M.G.; McArthur, A.G.; Poinar, H.N.; Wright, G.D. Capturing the resistome: A targeted capture method to reveal antibiotic resistance determinants in metagenomes. Antimicrob. Agents Chemother. 2019, 64, e01324-19. [Google Scholar] [CrossRef]
- Ferreira, I.; Lepuschitz, S.; Beisken, S.; Fiume, G.; Mrazek, K.; Frank, B.J.H.; Huber, S.; Knoll, M.A.; von Haeseler, A.; Materna, A.; et al. Culture-free detection of antibiotic resistance markers from native patient samples by hybridization capture sequencing. Microorganisms 2021, 9, 1672. [Google Scholar] [CrossRef]
- Allicock, O.M.; Guo, C.; Uhlemann, A.-C.; Whittier, S.; Chauhan, L.V.; Garcia, J.; Price, A.; Morse, S.S.; Mishra, N.; Briese, T.; et al. BacCapSeq: A platform for diagnosis and characterization of bacterial infections. mBio 2018, 9, e02007-18. [Google Scholar] [CrossRef]
- Slizovskiy, I.B.; Oliva, M.; Settle, J.K.; Zyskina, L.V.; Prosperi, M.; Boucher, C.; Noyes, N.R. Target-Enriched Long-Read Sequencing (TELSeq) contextualizes antimicrobial resistance genes in metagenomes. Microbiome 2022, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Leekitcharoenphon, P.; Johansson, M.H.K.; Munk, P.; Malorny, B.; Skarżyńska, M.; Wadepohl, K.; Moyano, G.; Hesp, A.; Veldman, K.T.; Bossers, A.; et al. Genomic evolution of antimicrobial resistance in Escherichia coli. Sci. Rep. 2021, 11, 15108. [Google Scholar] [CrossRef]
- Merker, M.; Tueffers, L.; Vallier, M.; Groth, E.E.; Sonnenkalb, L.; Unterweger, D.; Baines, J.F.; Niemann, S.; Schulenburg, H. Evolutionary approaches to combat antibiotic resistance: Opportunities and challenges for precision medicine. Front. Immunol. 2020, 11, 1938. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Norris, S.J. In vitro cultivation of the syphilis spirochete Treponema pallidum. Curr. Protoc. 2021, 1, e44. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Borges, V.; Antelo, M.; Pinheiro, M.; Nunes, A.; Azevedo, J.; Borrego, M.J.; Mendonça, J.; Carpinteiro, D.; Vieira, L.; et al. Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation. Nat. Microbiol. 2016, 2, 16190. [Google Scholar] [CrossRef] [PubMed]
- Dennis, T.P.W.; Mable, B.K.; Brunelle, B.; Devault, A.; Carter, R.W.; Ling, C.L.; Mmbaga, B.T.; Halliday, J.E.B.; Oravcova, K.; Forde, T.L. Target-enrichment sequencing yields valuable genomic data for challenging-to-culture bacteria of public health importance. Microb. Genom. 2022, 8, mgen000836. [Google Scholar] [CrossRef] [PubMed]
- Boruah, A.P.; Kroopnick, A.; Thakkar, R.; Wapniarski, A.E.; Kim, C.; Dugue, R.; Harrigan, E.; Lipkin, W.I.; Mishra, N.; Thakur, K.T. Application of VirCapSeq-VERT and BacCapSeq in the diagnosis of presumed and definitive neuroinfectious diseases. J. Neurovirol. 2023, 29, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Dickson, Z.W.; Hackenberger, D.; Kuch, M.; Marzok, A.; Banerjee, A.; Rossi, L.; Klowak, J.A.; Fox-Robichaud, A.; Mossmann, K.; Miller, M.S.; et al. Probe design for simultaneous, targeted capture of diverse metagenomic targets. Cell Rep. Methods 2021, 1, 100069. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Sounart, H.; Lázár, E.; Masarapu, Y.; Wu, J.; Várkonyi, T.; Glasz, T.; Kiss, A.; Borgström, E.; Hill, A.; Rezene, S.; et al. Dual spatially resolved transcriptomics for human host-pathogen colocalization studies in FFPE tissue sections. Genome Biol. 2023, 24, 237. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.; Jiao, L.; Wang, Z.; Li, W.; Geldsetzer, P.; Bärnighausen, T.; et al. Estimates and projections of the global economic cost of 29 cancers in 204 countries and territories from 2020 to 2050. JAMA Oncol. 2023, 9, 465–472. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Cerasuolo, A.; Starita, N.; Amiranda, S.; Bonelli, P.; Tuccillo, F.M.; Buonaguro, F.M.; Buonaguro, L.; Tornesello, A.L. Reactivation of telomerase reverse transcriptase expression in cancer: The role of TERT promoter mutations. Front. Cell Dev. Biol. 2023, 11, 1286683. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. An introduction to virus infections and human cancer. In Viruses and Human Cancer. Recent Results in Cancer Research; Wu, T.C., Chang, M.H., Jeang, K.T., Eds.; Springer: Cham, Switzerland, 2021; Volume 217, pp. 1–11. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Magrini, V.; Becker, N.; Armstrong, J.R.; Demeter, R.T.; Wylie, T.; Abel, H.J.; Pfeifer, J.D. Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J. Mol. Diagn. 2011, 13, 325–333. [Google Scholar] [CrossRef]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.-C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef]
- Wen, Y.; Xu, H.; Han, J.; Jin, R.; Chen, H. How does Epstein-Barr virus interact with other microbiomes in EBV-driven cancers? Front. Cell Infect. Microbiol. 2022, 12, 852066. [Google Scholar] [CrossRef]
- Kwok, H.; Wu, C.W.; Palser, A.L.; Kellam, P.; Sham, P.C.; Kwong, D.L.W.; Chiang, A.K.S. Genomic diversity of Epstein-Barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. J. Virol. 2014, 88, 10662–10672. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Zhang, S.; Yao, Y.; Xiang, T.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics 2019, 9, 1115–1124. [Google Scholar] [CrossRef]
- Fernandes, Q.; Merhi, M.; Raza, A.; Inchakalody, V.P.; Abdelouahab, N.; Zar Gul, A.R.; Uddin, S.; Dermime, S. Role of Epstein-Barr virus in the pathogenesis of head and neck cancers and its potential as an immunotherapeutic target. Front. Oncol. 2018, 8, 257. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein-Barr virus: How its lytic phase contributes to oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef]
- Han, S.; Tay, J.K.; Loh, C.J.L.; Chu, A.J.M.; Yeong, J.P.S.; Lim, C.M.; Toh, H.C. Epstein-Barr virus epithelial cancers—A comprehensive understanding to drive novel therapies. Front. Immunol. 2021, 12, 734293. [Google Scholar] [CrossRef] [PubMed]
- Nauclér, C.S.; Geisler, J.; Vetvik, K. The emerging role of human cytomegalovirus infection in human carcinogenesis: A review of current evidence and potential therapeutic implications. Oncotarget 2019, 10, 4333–4347. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.; Touma, J.; Rahbar, A.; Söderberg-Nauclér, C.; Vetvik, K. A review of the potential role of human cytomegalovirus (HCMV) infections in breast cancer carcinogenesis and abnormal immunity. Cancers 2019, 11, 1842. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G. Tumors and cytomegalovirus: An intimate interplay. Viruses 2022, 14, 812. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2016, 2, vew017. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [PubMed]
- Balegamire, S.J.; McClymont, E.; Croteau, A.; Dodin, P.; Gantt, S.; Besharati, A.A.; Renaud, C.; Mâsse, B.; Boucoiran, I. Prevalence, incidence, and risk factors associated with cytomegalovirus infection in healthcare and childcare worker: A systematic review and meta-analysis. Syst. Rev. 2022, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human cytomegalovirus genomes sequenced directly from clinical material: Variation, multiple-strain infection, recombination, and gene loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole-genome sequencing directly from clinical specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.M.; Musonda, K.G.; Escriva, E.; Njenga, M.; Agbueze, A.; Camiolo, S.; Davison, A.J.; Gompels, U.A. Multiple-strain infections of human cytomegalovirus with high genomic diversity are common in breast milk from human immunodeficiency virus-infected women in Zambia. J. Infect. Dis. 2019, 220, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Görzer, I.; Trajanoski, S.; Popow-Kraupp, T.; Puchhammer-Stöckl, E. Analysis of human cytomegalovirus strain populations in urine samples of newborns by ultra deep sequencing. J. Clin. Virol. 2015, 73, 101–104. [Google Scholar] [CrossRef]
- Mayer, B.T.; Krantz, E.M.; Swan, D.; Ferrenberg, J.; Simmons, K.; Selke, S.; Huang, M.-L.; Casper, C.; Corey, L.; Wald, A.; et al. Transient oral human cytomegalovirus infections indicate inefficient viral spread from very few initially infected cells. J. Virol. 2017, 91, e00380-17. [Google Scholar] [CrossRef]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Lin, C.-L.; Tseng, T.-C.; Kao, J.-H. What can we learn from hepatitis B virus clinical cohorts? Liver Int. 2015, 35 (Suppl. S1), 91–99. [Google Scholar] [CrossRef]
- Ward, J.W.; Hinman, A.R. What is needed to eliminate hepatitis B virus and hepatitis C virus as global health threats. Gastroenterology 2019, 156, 297–310. [Google Scholar] [CrossRef]
- Bonsall, D.; Ansari, M.A.; Ip, C.; Trebes, A.; Brown, A.; Klenerman, P.; Buck, D.; Consortium, S.-H.; Piazza, P.; Barnes, E.; et al. ve-SEQ: Robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Res 2015, 4, 1062. [Google Scholar] [CrossRef]
- Thomson, E.; Ip, C.L.C.; Badhan, A.; Christiansen, M.T.; Adamson, W.; Ansari, M.A.; Bibby, D.; Breuer, J.; Brown, A.; Bowden, R.; et al. Comparison of next-generation sequencing technologies for comprehensive assessment of full-length hepatitis C viral genomes. J. Clin. Microbiol. 2016, 54, 2470–2484. [Google Scholar] [CrossRef] [PubMed]
- Munyuza, C.; Ji, H.; Lee, E.R. Probe capture enrichment methods for HIV and HCV genome sequencing and drug resistance genotyping. Pathogens 2022, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Begg, T.J.A.; Schmidt, A.; Kocher, A.; Larmuseau, M.H.D.; Runfeldt, G.; Maier, P.A.; Wilson, J.D.; Barquera, R.; Maj, C.; Szolek, A.; et al. Genomic analyses of hair from Ludwig van Beethoven. Curr. Biol. 2023, 33, 1431–1447.e22. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Doorbar, J. The low-risk papillomaviruses. Virus Res. 2017, 231, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi, P.; Fani, M.; Rezayi, M.; Avan, A.; Pasdar, Z.; Karimi, E.; Amiri, I.S.; Ghayour-Mobarhan, M. Early detection of cervical cancer based on high-risk HPV DNA-based genosensors: A systematic review. Biofactors 2019, 45, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Unger, E.R.; Batra, D.; Sheth, M.; Steinau, M.; Jasinski, J.; Jones, J.; Rajeevan, M.S. Universal human papillomavirus typing assay: Whole-genome sequencing following target enrichment. J. Clin. Microbiol. 2017, 55, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Unger, E.R.; Rajeevan, M.S. Universal human papillomavirus typing by whole genome sequencing following target enrichment: Evaluation of assay reproducibility and limit of detection. BMC Genom. 2019, 20, 231. [Google Scholar] [CrossRef]
- McBride, A.A. Human papillomaviruses: Diversity, infection and host interactions. Nat. Rev. Microbiol. 2022, 20, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Du, J. Human papillomavirus vaccines: An updated review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Wang, R.; Pan, W.; Jin, L.; Huang, W.; Li, Y.; Wu, D.; Gao, C.; Ma, D.; Liao, S. Human papillomavirus vaccine against cervical cancer: Opportunity and challenge. Cancer Lett. 2020, 471, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Jani, C.; Al Omari, O.; Singh, H.; Walker, A.; Patel, K.; Mouchati, C.; Radwan, A.; Pandit, Z.; Hanbury, G.; Crowley, C.; et al. Trends of HIV-related cancer mortality between 2001 and 2018: An observational analysis. Trop. Med. Infect. Dis. 2021, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Head, M.G.; Brown, R.J.; Newell, M.-L.; Scott, J.A.G.; Batchelor, J.; Atun, R. The allocation of US$105 billion in global funding from G20 countries for infectious disease research between 2000 and 2017: A content analysis of investments. Lancet Glob. Health 2020, 8, e1295–e1304. [Google Scholar] [CrossRef] [PubMed]
- Pitman, M.C.; Lewin, S.R. Towards a cure for human immunodeficiency virus. Intern. Med. J. 2018, 48, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.; Kirchner, R.; Amr, S.S.; Mansur, L.; Shakhbatyan, R.; Kim, M.; Bosque, A.; Siliciano, R.F.; Planelles, V.; Hofmann, O.; et al. HIV integration site analysis of cellular models of hiv latency with a probe-enriched next-generation sequencing assay. J. Virol. 2016, 90, 4511–4519. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.C.; Miyazato, P.; Katsuya, H.; Islam, S.; Yang, B.T.J.; Ito, J.; Matsuo, M.; Takeuchi, H.; Ishida, T.; Matsuda, K.; et al. HIV-1 DNA-capture-seq is a useful tool for the comprehensive characterization of HIV-1 provirus. Sci. Rep. 2019, 9, 12326. [Google Scholar] [CrossRef]
- Colson, P.; Dhiver, C.; Tamalet, C.; Delerce, J.; Glazunova, O.O.; Gaudin, M.; Levasseur, A.; Raoult, D. Dramatic HIV DNA degradation associated with spontaneous HIV suppression and disease-free outcome in a young seropositive woman following her infection. Sci. Rep. 2020, 10, 2548. [Google Scholar] [CrossRef]
- Miyazato, P.; Katsuya, H.; Fukuda, A.; Uchiyama, Y.; Matsuo, M.; Tokunaga, M.; Hino, S.; Nakao, M.; Satou, Y. Application of targeted enrichment to next-generation sequencing of retroviruses integrated into the host human genome. Sci. Rep. 2016, 6, 28324. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal target capture of HIV sequences from NGS libraries. Front. Microbiol. 2018, 9, 2150. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Vallari, A.; McArthur, C.; Sthreshley, L.; Cloherty, G.A.; Berg, M.G.; Rodgers, M.A. Brief report: Complete genome sequence of CG-0018a-01 establishes HIV-1 subtype L. J. Acquir. Immune Defic. Syndr. 2020, 83, 319–322. [Google Scholar] [CrossRef]
- Bonsall, D.; Golubchik, T.; de Cesare, M.; Limbada, M.; Kosloff, B.; MacIntyre-Cockett, G.; Hall, M.; Wymant, C.; Ansari, M.A.; Abeler-Dörner, L.; et al. A comprehensive genomics solution for HIV surveillance and clinical monitoring in low-income settings. J. Clin. Microbiol. 2020, 58, e00382-20. [Google Scholar] [CrossRef]
- Katz, J.M.; Wang, M.; Webster, R.G. Direct sequencing of the HA gene of influenza (H3N2) virus in original clinical samples reveals sequence identity with mammalian cell-grown virus. J. Virol. 1990, 64, 1808–1811. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Tang, J.W.-T.; Kong, D.H.-L.; Loh, T.P.; Chiang, D.K.-L.; Lam, T.T.-Y.; Koay, E.S.-C. Comparison of mutation patterns in full-genome A/H3N2 influenza sequences obtained directly from clinical samples and the same samples after a single MDCK passage. PLoS ONE 2013, 8, e79252. [Google Scholar] [CrossRef] [PubMed]
- Aguilar Rangel, M.; Dolan, P.T.; Taguwa, S.; Xiao, Y.; Andino, R.; Frydman, J. High-resolution mapping reveals the mechanism and contribution of genome insertions and deletions to RNA virus evolution. Proc. Natl. Acad. Sci. USA 2023, 120, e2304667120. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Lee, K.S.; Beach, A.; Sanford, B.; Baird, N.L.; Como, C.; Graybill, C.; Jones, D.; Tekeste, E.; Ballard, M.; et al. Targeted genome sequencing reveals varicella-zoster virus open reading frame 12 deletion. J. Virol. 2017, 91, e01141-17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Liao, W.-C.; Ger, L.-P.; Chen, J.-C.; Hsu, T.-I.; Lee, Y.-C.; Chang, H.-T.; Chen, Y.-C.; Jan, Y.-H.; Lee, K.-H.; et al. Carboxyl-terminal modulator protein positively regulates Akt phosphorylation and acts as an oncogenic driver in breast cancer. Cancer Res. 2013, 73, 6194–6205. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Houff, S. Neurological complications of herpes simplex virus type 2 infection. Arch. Neurol. 2008, 65, 596–600. [Google Scholar] [CrossRef]
- Greninger, A.L.; Roychoudhury, P.; Xie, H.; Casto, A.; Cent, A.; Pepper, G.; Koelle, D.M.; Huang, M.-L.; Wald, A.; Johnston, C.; et al. Ultrasensitive capture of human herpes simplex virus genomes directly from clinical samples reveals extraordinarily limited evolution in cell culture. mSphere 2018, 3, e00283-18. [Google Scholar] [CrossRef]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef]
- Renner, D.W.; Szpara, M.L. Impacts of genome-wide analyses on our understanding of human herpesvirus diversity and evolution. J. Virol. 2018, 92, e00908-17. [Google Scholar] [CrossRef]
- Chiang, D.A.; Dekker, J.P. From the pipeline to the bedside: Advances and challenges in clinical metagenomics. J. Infect. Dis. 2020, 221, S331–S340. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Mielonen, O.I.; Pratas, D.; Hedman, K.; Sajantila, A.; Perdomo, M.F. Detection of low-copy human virus DNA upon prolonged formalin fixation. Viruses 2022, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Toppinen, M.; Pratas, D.; Väisänen, E.; Söderlund-Venermo, M.; Hedman, K.; Perdomo, M.F.; Sajantila, A. The landscape of persistent human DNA viruses in femoral bone. Forensic Sci. Int. Genet. 2020, 48, 102353. [Google Scholar] [CrossRef] [PubMed]
- Nabel, C.S.; Sameroff, S.; Shilling, D.; Alapat, D.; Ruth, J.R.; Kawano, M.; Sato, Y.; Stone, K.; Spetalen, S.; Valdivieso, F.; et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric Castleman disease. PLoS ONE 2019, 14, e0218660. [Google Scholar] [CrossRef] [PubMed]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.A.; Nijhuis, W.; Leavis, H.L.; Riezebos-Brilman, A.; Lindemans, C.A.; Schuurman, R. Broad virus detection and variant discovery in fecal samples of hematopoietic transplant recipients using targeted sequence capture metagenomics. Front. Microbiol. 2020, 11, 560179. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Roy, S.; Ruis, C.; Yara Romero, E.; Shah, D.; Williams, R.; Breuer, J. Norovirus whole-genome sequencing by SureSelect target enrichment: A robust and sensitive method. J. Clin. Microbiol. 2016, 54, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, S.; Gorham, J.M.; Mazaika, E.; Parfenov, M.; Dang, X.; DePalma, S.; McKean, D.; Seidman, C.E.; Seidman, J.G.; Koralnik, I.J. ViroFind: A novel target-enrichment deep-sequencing platform reveals a complex JC virus population in the brain of PML patients. PLoS ONE 2018, 13, e0186945. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 2019, 37, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, A.; Mukerji, S.S.; Ye, S.; Leone, M.J.; Freimark, L.M.; Park, D.; Adams, G.; Lemieux, J.; Kanjilal, S.; Solomon, I.H.; et al. Enhanced virus detection and metagenomic sequencing in patients with meningitis and encephalitis. mBio 2021, 12, e0114321. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, V.; Briese, T.; Ranjan, A.; Donovan, W.M.; Mansukhani, M.M.; Chowdhary, R.; Lipkin, W.I. Validation of the VirCapSeq-VERT system for differential diagnosis, detection, and surveillance of viral infections. J. Clin. Microbiol. 2024, 62, e00612-23. [Google Scholar] [CrossRef]
- Cummings, M.J.; Tokarz, R.; Bakamutumaho, B.; Kayiwa, J.; Byaruhanga, T.; Owor, N.; Namagambo, B.; Wolf, A.; Mathema, B.; Lutwama, J.J.; et al. Precision surveillance for viral respiratory pathogens: Virome capture sequencing for the detection and genomic characterization of severe acute respiratory infection in Uganda. Clin. Infect. Dis. 2019, 68, 1118–1125. [Google Scholar] [CrossRef]
- Kim, K.W.; Deveson, I.W.; Pang, C.N.I.; Yeang, M.; Naing, Z.; Adikari, T.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Verich, A.; et al. Respiratory viral co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Sci. Rep. 2021, 11, 3934. [Google Scholar] [CrossRef]
- Pogka, V.; Papadopoulou, G.; Valiakou, V.; Sgouras, D.N.; Mentis, A.F.; Karamitros, T. Targeted virome sequencing enhances unbiased detection and genome assembly of known and emerging viruses—The example of SARS-CoV-2. Viruses 2022, 14, 1272. [Google Scholar] [CrossRef] [PubMed]
- McGill, F.; Tokarz, R.; Thomson, E.C.; Filipe, A.; Sameroff, S.; Jain, K.; Bhuva, N.; Ashraf, S.; Lipkin, W.I.; Corless, C.; et al. Viral capture sequencing detects unexpected viruses in the cerebrospinal fluid of adults with meningitis. J. Infect. 2022, 84, 499–510. [Google Scholar] [CrossRef]
- Williams, S.H.; Cordey, S.; Bhuva, N.; Laubscher, F.; Hartley, M.-A.; Boillat-Blanco, N.; Mbarack, Z.; Samaka, J.; Mlaganile, T.; Jain, K.; et al. Investigation of the plasma virome from cases of unexplained febrile illness in Tanzania from 2013 to 2014: A comparative analysis between unbiased and VirCapSeq-VERT high-throughput sequencing approaches. mSphere 2018, 3, 00311-18. [Google Scholar] [CrossRef]
- Batool, M.; Galloway-Peña, J. Clinical metagenomics-challenges and future prospects. Front. Microbiol. 2023, 14, 1186424. [Google Scholar] [CrossRef] [PubMed]
- Launes, C.; Camacho, J.; Pons-Espinal, M.; López-Labrador, F.X.; Esteva, C.; Cabrerizo, M.; Fernández-García, M.D.; Fogeda, M.; Masa-Calles, J.; López-Perea, N.; et al. Hybrid capture shotgun sequencing detected unexpected viruses in the cerebrospinal fluid of children with acute meningitis and encephalitis. Eur. J. Clin. Microbiol. Infect. Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Plasmodium—A brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Anthropol. 2021, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Strategic Advisory Group on Malaria Eradication. Malaria Eradication: Benefits, Future Scenarios and Feasibility. A Report of the Strategic Advisory Group on Malaria Eradication; World Health Organization: Geneva, Switzerland, 2020; pp. 4–71. [Google Scholar]
- Battle, K.E.; Lucas, T.C.D.; Nguyen, M.; Howes, R.E.; Nandi, A.K.; Twohig, K.A.; Pfeffer, D.A.; Cameron, E.; Rao, P.C.; Casey, D.; et al. Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000–17: A spatial and temporal modelling study. Lancet 2019, 394, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.J.; Lucas, T.C.D.; Nguyen, M.; Nandi, A.K.; Bisanzio, D.; Battle, K.E.; Cameron, E.; Twohig, K.A.; Pfeffer, D.A.; Rozier, J.A.; et al. Mapping the global prevalence, incidence, and mortality of Plasmodium falciparum, 2000–17: A spatial and temporal modelling study. Lancet 2019, 394, 322–331. [Google Scholar] [CrossRef]
- Melnikov, A.; Galinsky, K.; Rogov, P.; Fennell, T.; Van Tyne, D.; Russ, C.; Daniels, R.; Barnes, K.G.; Bochicchio, J.; Ndiaye, D.; et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011, 12, R73. [Google Scholar] [CrossRef]
- Smith, M.; Campino, S.; Gu, Y.; Clark, T.G.; Otto, T.D.; Maslen, G.; Manske, M.; Imwong, M.; Dondorp, A.M.; Kwiatkowski, D.P.; et al. An in-solution hybridisation method for the isolation of pathogen DNA from human DNA-rich clinical samples for analysis by NGS. Open Genom. J. 2012, 5, 18–29. [Google Scholar] [CrossRef]
- Bright, A.T.; Tewhey, R.; Abeles, S.; Chuquiyauri, R.; Llanos-Cuentas, A.; Ferreira, M.U.; Schork, N.J.; Vinetz, J.M.; Winzeler, E.A. Whole genome sequencing analysis of Plasmodium vivax using whole genome capture. BMC Genom. 2012, 13, 262. [Google Scholar] [CrossRef]
- Gural, N.; Mancio-Silva, L.; Miller, A.B.; Galstian, A.; Butty, V.L.; Levine, S.S.; Patrapuvich, R.; Desai, S.P.; Mikolajczak, S.A.; Kappe, S.H.I.; et al. In vitro culture, drug sensitivity, and transcriptome of Plasmodium vivax hypnozoites. Cell Host Microbe 2018, 23, 395–406.e4. [Google Scholar] [CrossRef]
- Pisarski, K. The global burden of disease of zoonotic parasitic diseases: Top 5 contenders for priority consideration. Trop. Med. Infect. Dis. 2019, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Cable, J.; Barber, I.; Boag, B.; Ellison, A.R.; Morgan, E.R.; Murray, K.; Pascoe, E.L.; Sait, S.M.; Wilson, A.J.; Booth, M. Global change, parasite transmission and disease control: Lessons from ecology. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160088. [Google Scholar] [CrossRef]
- Chevalier, F.D.; Valentim, C.L.; LoVerde, P.T.; Anderson, T.J. Efficient linkage mapping using exome capture and extreme QTL in schistosome parasites. BMC Genom. 2014, 15, 617. [Google Scholar] [CrossRef]
- Le Clec’h, W.; Chevalier, F.D.; Mattos, A.C.A.; Strickland, A.; Diaz, R.; McDew-White, M.; Rohr, C.M.; Kinung’hi, S.; Allan, F.; Webster, B.L.; et al. Genetic analysis of praziquantel response in schistosome parasites implicates a transient receptor potential channel. Sci. Transl. Med. 2021, 13, eabj9114. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, F.D.; Le Clec’h, W.; McDew-White, M.; Menon, V.; Guzman, M.A.; Holloway, S.P.; Cao, X.; Taylor, A.B.; Kinung’hi, S.; Gouvras, A.N.; et al. Oxamniquine resistance alleles are widespread in old world Schistosoma mansoni and predate drug deployment. PLoS Pathog. 2019, 15, e1007881. [Google Scholar] [CrossRef] [PubMed]
- Le Clec’h, W.; Chevalier, F.D.; McDew-White, M.; Allan, F.; Webster, B.L.; Gouvras, A.N.; Kinunghi, S.; Tchuem Tchuenté, L.-A.; Garba, A.; Mohammed, K.A.; et al. Whole genome amplification and exome sequencing of archived schistosome miracidia. Parasitology 2018, 145, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.A. Virulence factors of schistosomes. Microbes Infect. 2012, 14, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Hambrook, J.R.; Kaboré, A.L.; Pila, E.A.; Hanington, P.C. A metalloprotease produced by larval Schistosoma mansoni facilitates infection establishment and maintenance in the snail host by interfering with immune cell function. PLoS Pathog. 2018, 14, e1007393. [Google Scholar] [CrossRef]
- Bern, C.; Maguire, J.H.; Alvar, J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2008, 2, e313. [Google Scholar] [CrossRef]
- Bailey, F.; Mondragon-Shem, K.; Hotez, P.; Ruiz-Postigo, J.A.; Al-Salem, W.; Acosta-Serrano, Á.; Molyneux, D.H. A new perspective on cutaneous leishmaniasis—Implications for global prevalence and burden of disease estimates. PLoS Negl. Trop. Dis. 2017, 11, e0005739. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Revill, P.; Malvolti, S.; Malhame, M.; Sculpher, M.; Kaye, P.M. Estimating the global demand curve for a leishmaniasis vaccine: A generalisable approach based on global burden of disease estimates. PLoS Negl. Trop. Dis. 2022, 16, e0010471. [Google Scholar] [CrossRef]
- Kima, P.E. The amastigote forms of Leishmania are experts at exploiting host cell processes to establish infection and persist. Int. J. Parasitol. 2007, 37, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Lypaczeeski, P.; Johanna, H.; Zhang, W.-W.; McCall, L.-I.; Torcivia-Rodriguez, J.; Simonyan, V.; Kaur, K.; Dewar, K.; Matlashewski, G. A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci. Rep. 2018, 8, 16549. [Google Scholar] [CrossRef]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, I.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [PubMed]
- Lypaczewski, P.; Thakur, L.; Jain, A.; Kumari, S.; Paulini, K.; Matlashewski, G.; Jain, M. An intraspecies Leishmania donovani hybrid from the Indian subcontinent is associated with an atypical phenotype of cutaneous disease. iScience 2022, 25, 103802. [Google Scholar] [CrossRef]
- Camacho, E.; González-de la Fuente, S.; Rastrojo, A.; Peiró-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Imamura, H.; Sanders, M.; Broeck, F.V.d.; Bhattarai, N.R.; Vanaerschot, M.; Maes, I.; D’Haenens, E.; Rai, K.; Rijal, S.; et al. Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PLoS Negl. Trop. Dis. 2019, 13, e0007900. [Google Scholar] [CrossRef]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. mBio 2017, 8, e00599-17. [Google Scholar] [CrossRef]
- Lopez, L.; Koepfli, C. Systematic review of Plasmodium falciparum and Plasmodium vivax polyclonal infections: Impact of prevalence, study population characteristics, and laboratory procedures. PLoS ONE 2021, 16, e0249382. [Google Scholar] [CrossRef]
- Gannavaram, S.; Torcivia, J.; Gasparyan, L.; Kaul, A.; Ismail, N.; Simonyan, V.; Nakhasi, H.L. Whole genome sequencing of live attenuated Leishmania donovani parasites reveals novel biomarkers of attenuation and enables product characterization. Sci. Rep. 2017, 7, 4718. [Google Scholar] [CrossRef]
- Moser, K.A.; Drábek, E.F.; Dwivedi, A.; Stucke, E.M.; Crabtree, J.; Dara, A.; Shah, Z.; Adams, M.; Li, T.; Rodrigues, P.T.; et al. Strains used in whole organism Plasmodium falciparum vaccine trials differ in genome structure, sequence, and immunogenic potential. Genome Med. 2020, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.C.; Phelan, J.; Francisco, A.F.; Taylor, M.C.; Lewis, M.D.; Pain, A.; Clark, T.G.; Kelly, J.M. Genome-wide mutagenesis and multi-drug resistance in American trypanosomes induced by the front-line drug benznidazole. Sci. Rep. 2017, 7, 14407. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, S.; Kubota, R.; Nishi, T.; Kamchonwongpaisan, S.; Srichairatanakool, S.; Shinzawa, N.; Syafruddin, D.; Yuda, M.; Uthaipibull, C. Genome-wide functional screening of drug-resistance genes in Plasmodium falciparum. Nat. Commun. 2022, 13, 6163. [Google Scholar] [CrossRef] [PubMed]
- Crellen, T.; Allan, F.; David, S.; Durrant, C.; Huckvale, T.; Holroyd, N.; Emery, A.M.; Rollinson, D.; Aanensen, D.M.; Berriman, M.; et al. Whole genome resequencing of the human parasite Schistosoma mansoni reveals population history and effects of selection. Sci. Rep. 2016, 6, 20954. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.N.; Le Clec’h, W.; Chevalier, F.D.; McDew-White, M.; LoVerde, P.T.; Ramiro de Assis, R.; Oliveira, G.; Kinung’hi, S.; Djirmay, A.G.; Steinauer, M.L.; et al. Genomic analysis of a parasite invasion: Colonization of the Americas by the blood fluke Schistosoma mansoni. Mol. Ecol. 2022, 31, 2242–2263. [Google Scholar] [CrossRef] [PubMed]
- Head, M.G.; Goss, S.; Gelister, Y.; Alegana, V.; Brown, R.J.; Clarke, S.C.; Fitchett, J.R.A.; Atun, R.; Scott, J.A.G.; Newell, M.-L.; et al. Global funding trends for malaria research in sub-Saharan Africa: A systematic analysis. Lancet Glob. Health 2017, 5, e772–e781. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, Z.A.; Sommerfeld, J.; Lassi, Z.S.; Salam, R.A.; Das, J.K. Global burden, distribution, and interventions for infectious diseases of poverty. Infect. Dis. Poverty 2014, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Ochola, E.A.; Karanja, D.M.S.; Elliott, S.J. The impact of neglected tropical diseases (NTDs) on health and wellbeing in sub-Saharan Africa (SSA): A case study of Kenya. PLoS Negl. Trop. Dis. 2021, 15, e0009131. [Google Scholar] [CrossRef]
- Tigabu, A.; Taye, S.; Aynalem, M.; Adane, K. Prevalence and associated factors of intestinal parasitic infections among patients attending Shahura Health Center, Northwest Ethiopia. BMC Res. Notes 2019, 12, 333. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Alvarez, I.; Goyens, P.; Luizaga-López, J.M.; Jacobs, F. Geographic differences in the distribution of parasitic infections in children of Bolivia. Parasite Epidemiol. Control 2021, 14, e00217. [Google Scholar] [CrossRef]
- Martviset, P.; Phadungsil, W.; Na-Bangchang, K.; Sungkhabut, W.; Panupornpong, T.; Prathaphan, P.; Torungkitmangmi, N.; Chaimon, S.; Wangboon, C.; Jamklang, M.; et al. Current prevalence and geographic distribution of helminth infections in the parasitic endemic areas of rural Northeastern Thailand. BMC Public Health 2023, 23, 448. [Google Scholar] [CrossRef] [PubMed]
- Bodimeade, C.; Marks, M.; Mabey, D. Neglected tropical diseases: Elimination and eradication. Clin. Med. 2019, 19, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Aya Pastrana, N.; Beran, D.; Somerville, C.; Heller, O.; Correia, J.C.; Suggs, L.S. The process of building the priority of neglected tropical diseases: A global policy analysis. PLoS Negl. Trop. Dis. 2020, 14, e0008498. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J. Human parasitology and parasitic diseases: Heading towards 2050. Adv. Parasitol. 2018, 100, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Mejia, A.M.; Hall, B.S.; Taylor, M.C.; Gómez-Palacio, A.; Wilkinson, S.R.; Triana-Chávez, O.; Kelly, J.M. Benznidazole-resistance in Trypanosoma cruzi is a readily acquired trait that can arise independently in a single population. J. Infect. Dis. 2012, 206, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Amorim-Vaz, S.; Tran, V.D.T.; Pradervand, S.; Pagni, M.; Coste, A.T.; Sanglard, D. RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans. mBio 2015, 6, e00942-15. [Google Scholar] [CrossRef]
- Schrevens, S.; Durandau, E.; Tran, V.D.T.; Sanglard, D. Using in vivo transcriptomics and RNA enrichment to identify genes involved in virulence of Candida glabrata. Virulence 2022, 13, 1285–1303. [Google Scholar] [CrossRef]
- Hovhannisyan, H.; Rodríguez, A.; Saus, E.; Vaneechoutte, M.; Gabaldón, T. Multiplexed target enrichment of coding and non-coding transcriptomes enables studying Candida spp. infections from human derived samples. Front. Cell Infect. Microbiol. 2023, 13, 1093178. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef]
- Bartoszewicz, J.M.; Nasri, F.; Nowicka, M.; Renard, B.Y. Detecting DNA of novel fungal pathogens using ResNets and a curated fungi-hosts data collection. Bioinformatics 2022, 38, ii168–ii174. [Google Scholar] [CrossRef] [PubMed]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and multi-national prevalence of fungal diseases-estimate precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef]
- Firacative, C. Invasive fungal disease in humans: Are we aware of the real impact? Mem. Inst. Oswaldo Cruz 2020, 115, e200430. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nosanchuk, J.D. Fungal diseases as neglected pathogens: A wake-up call to public health officials. PLoS Negl. Trop. Dis. 2020, 14, e0007964. [Google Scholar] [CrossRef]
- Rahman, M.T.; Sobur, M.A.; Islam, M.S.; Ievy, S.; Hossain, M.J.; El Zowalaty, M.E.; Rahman, A.T.; Ashour, H.M. Zoonotic diseases: Etiology, impact, and control. Microorganisms 2020, 8, 1405. [Google Scholar] [CrossRef] [PubMed]
- Leal Filho, W.; Ternova, L.; Parasnis, S.A.; Kovaleva, M.; Nagy, G.J. Climate change and zoonoses: A review of concepts, definitions, and bibliometrics. Int. J. Environ. Res. Public Health 2022, 19, 893. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Sullivan, N.J. Emerging viral diseases from a vaccinology perspective: Preparing for the next pandemic. Nat. Immunol. 2018, 19, 20–28. [Google Scholar] [CrossRef]
- Roychoudhury, S.; Das, A.; Sengupta, P.; Dutta, S.; Roychoudhury, S.; Choudhury, A.P.; Ahmed, A.B.F.; Bhattacharjee, S.; Slama, P. Viral pandemics of the last four decades: Pathophysiology, health impacts and perspectives. Int. J. Environ. Res. Public Health 2020, 17, 9411. [Google Scholar] [CrossRef]
- Bhadoria, P.; Gupta, G.; Agarwal, A. Viral pandemics in the past two decades: An overview. J. Family Med. Prim. Care 2021, 10, 2745–2750. [Google Scholar] [CrossRef]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.-F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef]
- Tajudeen, Y.A.; Oladipo, H.J.; Oladunjoye, I.O.; Mustapha, M.O.; Mustapha, S.T.; Abdullahi, A.A.; Yusuf, R.O.; Abimbola, S.O.; Adebayo, A.O.; Ikebuaso, J.G.; et al. Preventing the next pandemic through a planetary health approach: A focus on key drivers of zoonosis. Challenges 2022, 13, 50. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2019, 4, 10–19. [Google Scholar] [CrossRef]
- Carroll, D.; Morzaria, S.; Briand, S.; Johnson, C.K.; Morens, D.; Sumption, K.; Tomori, O.; Wacharphaueasadee, S. Preventing the next pandemic: The power of a global viral surveillance network. BMJ 2021, 372, n485. [Google Scholar] [CrossRef]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]
- Cable, J.; Fauci, A.; Dowling, W.E.; Günther, S.; Bente, D.A.; Yadav, P.D.; Madoff, L.C.; Wang, L.-F.; Arora, R.K.; Van Kerkhove, M.; et al. Lessons from the pandemic: Responding to emerging zoonotic viral diseases—A keystone symposia report. Ann. N. Y. Acad. Sci. 2022, 1518, 209–225. [Google Scholar] [CrossRef]
- Aryaprema, V.S.; Steck, M.R.; Peper, S.T.; Xue, R.; Qualls, W.A. A systematic review of published literature on mosquito control action thresholds across the world. PLoS Negl. Trop. Dis. 2023, 17, e0011173. [Google Scholar] [CrossRef] [PubMed]
- Ronca, S.E.; Ruff, J.C.; Murray, K.O. A 20-year historical review of West Nile virus since its initial emergence in North America: Has West Nile virus become a neglected tropical disease? PLoS Negl. Trop. Dis. 2021, 15, e0009190. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R. Epidemiology of West Nile virus in the United States: Implications for arbovirology and public health. J. Med. Entomol. 2019, 56, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.; Mathis, S.; Martin, S.W.; Erin Staples, J.; Fischer, M.; Lindsey, N.P. Surveillance for West Nile virus disease—United States, 2009–2018. Am. J. Transplant. 2021, 21, 1959–1974. [Google Scholar] [CrossRef]
- Naccache, S.N.; Thézé, J.; Sardi, S.I.; Somasekar, S.; Greninger, A.L.; Bandeira, A.C.; Campos, G.S.; Tauro, L.B.; Faria, N.R.; Pybus, O.G.; et al. Distinct Zika virus lineage in Salvador, Bahia, Brazil. Emerg. Infect. Dis. 2016, 22, 1788–1792. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Kraemer, M.U.G.; Dudas, G.; Tan, A.L.; Gangavarapu, K.; Wiley, M.R.; White, S.; Thézé, J.; Magnani, D.M.; et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 2017, 546, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Li, T.; du Plessis, L.; Bouquet, J.; Kraemer, M.U.G.; Somasekar, S.; Yu, G.; de Cesare, M.; Balmaseda, A.; Kuan, G.; et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe 2018, 23, 855–864.e7. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Grubaugh, N.D.; Pybus, O.G.; Andersen, K.G. Precision epidemiology for infectious disease control. Nat. Med. 2019, 25, 206–211. [Google Scholar] [CrossRef]
- Kamaraj, U.S.; Tan, J.H.; Mei, O.X.; Pan, L.; Chawla, T.; Uehara, A.; Wang, L.-F.; Ooi, E.E.; Gubler, D.J.; Tissera, H.; et al. Application of a targeted-enrichment methodology for full-genome sequencing of dengue 1–4, chikungunya and Zika viruses directly from patient samples. PLoS Negl. Trop. Dis. 2019, 13, e0007184. [Google Scholar] [CrossRef] [PubMed]
- Madison-Antenucci, S.; Kramer, L.D.; Gebhardt, L.L.; Kauffman, E. Emerging tick-borne diseases. Clin. Microbiol. Rev. 2020, 33, e00083-18. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.S.; Hart, C.E.; Hermance, M.E.; Brining, D.L.; Thangamani, S. An overview of animal models for arthropod-borne viruses. Comp. Med. 2017, 67, 232–241. [Google Scholar] [PubMed]
- Rochlin, I.; Toledo, A. Emerging tick-borne pathogens of public health importance: A mini-review. J. Med. Microbiol. 2020, 69, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Tagliafierro, T.; Marques, A.; Sanchez-Vicente, S.; Gokden, A.; Fallon, B.; Mishra, N.; Briese, T.; Kapoor, V.; Sameroff, S.; et al. Development of a capture sequencing assay for enhanced detection and genotyping of tick-borne pathogens. Sci. Rep. 2021, 11, 12384. [Google Scholar] [CrossRef]
- Fatmi, S.S.; Zehra, R.; Carpenter, D.O. Powassan virus—A new reemerging tick-borne disease. Front. Public Health 2017, 5, 342. [Google Scholar] [CrossRef]
- Sanchez-Vicente, S.; Jain, K.; Tagliafierro, T.; Gokden, A.; Kapoor, V.; Guo, C.; Horn, E.J.; Lipkin, W.I.; Tokarz, R. Capture sequencing enables sensitive detection of tick-borne agents in human blood. Front. Microbiol. 2022, 13, 837621. [Google Scholar] [CrossRef]
- Aloke, C.; Obasi, N.A.; Aja, P.M.; Emelike, C.U.; Egwu, C.O.; Jeje, O.; Edeogu, C.O.; Onisuru, O.O.; Orji, O.U.; Achilonu, I. Combating Lassa fever in West African sub-region: Progress, challenges, and future perspectives. Viruses 2023, 15, 146. [Google Scholar] [CrossRef]
- Balogun, O.O.; Akande, O.W.; Hamer, D.H. Lassa fever: An evolving emergency in West Africa. Am. J. Trop. Med. Hyg. 2020, 104, 466–473. [Google Scholar] [CrossRef]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef]
- Oguzie, J.U.; Petros, B.A.; Oluniyi, P.E.; Mehta, S.B.; Eromon, P.E.; Nair, P.; Adewale-Fasoro, O.; Ifoga, P.D.; Odia, I.; Pastusiak, A.; et al. Metagenomic surveillance uncovers diverse and novel viral taxa in febrile patients from Nigeria. Nat. Commun. 2023, 14, 4693. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.T.; Crozier, I.; Fischer, W.A.; Hewlett, A.; Kraft, C.S.; Vega, M.-A.d.L.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola virus disease. Nat. Rev. Dis. Primers 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.-J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 972. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Jia, N.; Zhang, Y.-W.; Shum, M.H.-H.; Jiang, J.-F.; Zhu, H.-C.; Tong, Y.-G.; Shi, Y.-X.; Ni, X.-B.; Liao, Y.-S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef]
- Peng, M.-S.; Li, J.-B.; Cai, Z.-F.; Liu, H.; Tang, X.; Ying, R.; Zhang, J.-N.; Tao, J.-J.; Yin, T.-T.; Zhang, T.; et al. The high diversity of SARS-CoV-2-related coronaviruses in pangolins alerts potential ecological risks. Zool. Res. 2021, 42, 834–844. [Google Scholar] [CrossRef]
- Shi, W.; Shi, M.; Que, T.-C.; Cui, X.-M.; Ye, R.-Z.; Xia, L.-Y.; Hou, X.; Zheng, J.-J.; Jia, N.; Xie, X.; et al. Trafficked Malayan pangolins contain viral pathogens of humans. Nat. Microbiol. 2022, 7, 1259–1269. [Google Scholar] [CrossRef]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.F.; Lee, C.B.; Pascoe, S.M.; How, C.B.; Chan, S.; Tan, J.H.; Yang, X.; Zhou, P.; Shi, Z.; Sessions, O.M.; et al. Detection and characterization of a novel bat-borne coronavirus in Singapore using multiple molecular approaches. J. Gen. Virol. 2019, 100, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Si, H.-R.; Zhu, Y.; Yang, X.-L.; Anderson, D.E.; Shi, Z.-L.; Wang, L.-F.; Zhou, P. Discovery of bat coronaviruses through surveillance and probe capture-based next-generation sequencing. mSphere 2020, 5, 00807-19. [Google Scholar] [CrossRef] [PubMed]
- Paskey, A.C.; Ng, J.H.J.; Rice, G.K.; Chia, W.N.; Philipson, C.W.; Foo, R.J.H.; Cer, R.Z.; Long, K.A.; Lueder, M.R.; Lim, X.F.; et al. Detection of recombinant Rousettus bat coronavirus GCCDC1 in lesser dawn bats (Eonycteris spelaea) in Singapore. Viruses 2020, 12, 539. [Google Scholar] [CrossRef] [PubMed]
- Paskey, A.C.; Frey, K.G.; Schroth, G.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genom. 2019, 20, 155. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Loos, K.D.; Suchan, D.M.; Russell, J.N.; Sies, A.N.; Kumakamba, C.; Muyembe, F.; Mbala Kingebeni, P.; Ngay Lukusa, I.; N’Kawa, F.; et al. Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats. eLife 2022, 11, e79777. [Google Scholar] [CrossRef]
- Carbo, E.C.; Sidorov, I.A.; Zevenhoven-Dobbe, J.C.; Snijder, E.J.; Claas, E.C.; Laros, J.F.J.; Kroes, A.C.M.; de Vries, J.J.C. Coronavirus discovery by metagenomic sequencing: A tool for pandemic preparedness. J. Clin. Virol. 2020, 131, 104594. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Puchol, S.; Itarte, M.; Rusiñol, M.; Forés, E.; Mejías-Molina, C.; Andrés, C.; Antón, A.; Quer, J.; Abril, J.F.; Girones, R.; et al. Exploring the diversity of coronavirus in sewage during COVID-19 pandemic: Don’t miss the forest for the trees. Sci. Total Environ. 2021, 800, 149562. [Google Scholar] [CrossRef]
- Wylezich, C.; Calvelage, S.; Schlottau, K.; Ziegler, U.; Pohlmann, A.; Höper, D.; Beer, M. Next-generation diagnostics: Virus capture facilitates a sensitive viral diagnosis for epizootic and zoonotic pathogens including SARS-CoV-2. Microbiome 2021, 9, 51. [Google Scholar] [CrossRef]
- Arita, I.; Jezek, Z.; Khodakevich, L.; Ruti, K. Human monkeypox: A newly emerged Orthopoxvirus zoonosis in the tropical rain forests of Africa. Am. J. Trop. Med. Hyg. 1985, 34, 781–789. [Google Scholar] [CrossRef]
- Khodakevich, L.; Jezek, Z.; Messinger, D. Monkeypox virus: Ecology and public health significance. Bull. World Health Organ. 1988, 66, 747–752. [Google Scholar]
- Patrono, L.V.; Pléh, K.; Samuni, L.; Ulrich, M.; Röthemeier, C.; Sachse, A.; Muschter, S.; Nitsche, A.; Couacy-Hymann, E.; Boesch, C.; et al. Monkeypox virus emergence in wild chimpanzees reveals distinct clinical outcomes and viral diversity. Nat. Microbiol. 2020, 5, 955–965. [Google Scholar] [CrossRef]
- Berthet, N.; Descorps-Declère, S.; Besombes, C.; Curaudeau, M.; Nkili Meyong, A.A.; Selekon, B.; Labouba, I.; Gonofio, E.C.; Ouilibona, R.S.; Simo Tchetgna, H.D.; et al. Genomic History of human monkey pox infections in the Central African Republic between 2001 and 2018. Sci. Rep. 2021, 11, 13085. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, P.; Sereewit, J.; Xie, H.; Nunley, E.; Bakhash, S.M.; Lieberman, N.A.P.; Greninger, A.L. Genomic analysis of early monkeypox virus outbreak strains, Washington, USA. Emerg. Infect. Dis. 2023, 29, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Han, B.A.; Kramer, A.M.; Drake, J.M. Global patterns of zoonotic disease in mammals. Trends Parasitol. 2016, 32, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Relman, D.A. Opinion: To stop the next pandemic, we need to unravel the origins of COVID-19. Proc. Natl. Acad. Sci. USA 2020, 117, 29246–29248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Eng. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Dhar Chowdhury, S.; Oommen, A.M. Epidemiology of COVID-19. J. Dig. Endosc. 2020, 11, 3–7. [Google Scholar] [CrossRef]
- Koelle, K.; Martin, M.A.; Antia, R.; Lopman, B.; Dean, N.E. The changing epidemiology of SARS-CoV-2. Science 2022, 375, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kang, L.; Shen, Z.; Li, X.; Wu, W.; Ma, W.; Fang, C.; Yang, F.; Jiang, X.; Gong, S.; et al. Dynamics of severe acute respiratory syndrome coronavirus 2 genome variants in the feces during convalescence. J. Genet. Genom. 2020, 47, 610–617. [Google Scholar] [CrossRef]
- Rehn, A.; Braun, P.; Knüpfer, M.; Wölfel, R.; Antwerpen, M.H.; Walter, M.C. Catching SARS-CoV-2 by sequence hybridization: A comparative analysis. mSystems 2021, 6, e0039221. [Google Scholar] [CrossRef] [PubMed]
- Afiahayati; Bernard, S.; Gunadi; Wibawa, H.; Hakim, M.S.; Marcellus; Parikesit, A.A.; Dewa, C.K.; Sakakibara, Y. A comparison of bioinformatics pipelines for enrichment Illumina next generation sequencing systems in detecting SARS-CoV-2 virus strains. Genes 2022, 13, 1330. [Google Scholar] [CrossRef]
- Carbo, E.C.; Mourik, K.; Boers, S.A.; Munnink, B.O.; Nieuwenhuijse, D.; Jonges, M.; Welkers, M.R.A.; Matamoros, S.; van Harinxma Thoe Slooten, J.; Kraakman, M.E.M.; et al. A comparison of five Illumina, Ion Torrent, and nanopore sequencing technology-based approaches for whole genome sequencing of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 701–713. [Google Scholar] [CrossRef]
- Nicot, F.; Trémeaux, P.; Latour, J.; Jeanne, N.; Ranger, N.; Raymond, S.; Dimeglio, C.; Salin, G.; Donnadieu, C.; Izopet, J. Whole-genome sequencing of SARS-CoV-2: Comparison of target capture and amplicon single molecule real-time sequencing protocols. J. Med. Virol. 2023, 95, e28123. [Google Scholar] [CrossRef]
- Klempt, P.; Brož, P.; Kašný, M.; Novotný, A.; Kvapilová, K.; Kvapil, P. Performance of targeted library preparation solutions for SARS-CoV-2 whole genome analysis. Diagnostics 2020, 10, 769. [Google Scholar] [CrossRef] [PubMed]
- Ulhuq, F.R.; Barge, M.; Falconer, K.; Wild, J.; Fernandes, G.; Gallagher, A.; McGinley, S.; Sugadol, A.; Tariq, M.; Maloney, D.; et al. Analysis of the ARTIC V4 and V4.1 SARS-CoV-2 primers and their impact on the detection of Omicron BA.1 and BA.2 lineage-defining mutations. Microb. Genom. 2023, 9, mgen000991. [Google Scholar] [CrossRef] [PubMed]
- Kandel, S.; Hartzell, S.L.; Ingold, A.K.; Turner, G.A.; Kennedy, J.L.; Ussery, D.W. Genomic surveillance of SARS-CoV-2 using long-range PCR primers. Front. Microbiol. 2024, 15, 1272972. [Google Scholar] [CrossRef]
- Kreier, F. Deltacron: The story of the variant that wasn’t. Nature 2022, 602, 19. [Google Scholar] [CrossRef]
- Ceballos-Garzon, A.; Comtet-Marre, S.; Peyret, P. Applying targeted gene hybridization capture to viruses with a focus to SARS-CoV-2. Virus Res. 2023, 340, 199293. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Müller, N.F.; Wagner, C.; Frazar, C.D.; Roychoudhury, P.; Lee, J.; Moncla, L.H.; Pelle, B.; Richardson, M.; Ryke, E.; Xie, H.; et al. Viral genomes reveal patterns of the SARS-CoV-2 outbreak in Washington State. Sci. Transl. Med. 2021, 13, eabf0202. [Google Scholar] [CrossRef] [PubMed]
- Zrelovs, N.; Ustinova, M.; Silamikelis, I.; Birzniece, L.; Megnis, K.; Rovite, V.; Freimane, L.; Silamikele, L.; Ansone, L.; Pjalkovskis, J.; et al. First report on the Latvian SARS-CoV-2 isolate genetic diversity. Front. Med. 2021, 8, 626000. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, S.; Tang, Y.-P.; Zhang, S.; Xu, D.-Q.; Yue, S.-J.; Liu, Q.-L. Clinical Characteristics, Transmissibility, Pathogenicity, susceptible populations, and re-infectivity of prominent COVID-19 variants. Aging Dis. 2022, 13, 402–422. [Google Scholar] [CrossRef]
- Bolze, A.; Luo, S.; White, S.; Cirulli, E.T.; Wyman, D.; Dei Rossi, A.; Machado, H.; Cassens, T.; Jacobs, S.; Schiabor Barrett, K.M.; et al. SARS-CoV-2 variant Delta rapidly displaced variant Alpha in the United States and led to higher viral loads. Cell Rep. Med. 2022, 3, 100564. [Google Scholar] [CrossRef]
- Liu, J.; Wei, H.; He, D. Differences in case-fatality-rate of emerging SARS-CoV-2 variants. Public Health Pract. 2023, 5, 100350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, N.; Song, Y.; Song, R.; Pan, Y.; Wang, L.; Yan, S.; Wang, Q.; Ma, S.; Wei, L.; et al. Phylogenomic tracing of asymptomatic transmission in a COVID-19 outbreak. Innovation 2021, 2, 100099. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Cornforth, D.M.; Dushoff, J.; Weitz, J.S. The time scale of asymptomatic transmission affects estimates of epidemic potential in the COVID-19 outbreak. Epidemics 2020, 31, 100392. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.; He, Q.; Pascual, M. Quantifying asymptomatic infection and transmission of COVID-19 in New York City using observed cases, serology, and testing capacity. Proc. Natl. Acad. Sci. USA 2021, 118, e2019716118. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Uppal, T.; Hartley, P.D.; Gorzalski, A.; Pandori, M.; Picker, M.A.; Verma, S.C.; Pagilla, K. Detecting SARS-CoV-2 variants in wastewater and their correlation with circulating variants in the communities. Sci. Rep. 2022, 12, 16141. [Google Scholar] [CrossRef] [PubMed]
- Rowan, N.J.; Moral, R.A. Disposable face masks and reusable face coverings as non-pharmaceutical interventions (NPIs) to prevent transmission of SARS-CoV-2 variants that cause coronavirus disease (COVID-19): Role of new sustainable NPI design innovations and predictive mathematical modelling. Sci. Total Environ. 2021, 772, 145530. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Uddin, S.; Behbehani, M.; Abdul Razzack, N.; Zakir, F.; Shajan, A. SARS-CoV-2 in hospital air as revealed by comprehensive respiratory viral panel sequencing. Infect. Prev. Pract. 2022, 4, 100199. [Google Scholar] [CrossRef] [PubMed]
- Klompas, M.; Milton, D.K.; Rhee, C.; Baker, M.A.; Leekha, S. Current insights into respiratory virus transmission and potential implications for infection control programs. Ann. Intern. Med. 2021, 174, 1710–1718. [Google Scholar] [CrossRef]
- Greninger, A.L.; Zerr, D.M. NGSocomial Infections: High-resolution views of hospital-acquired infections through genomic epidemiology. J. Pediatr. Infect. Dis. Soc. 2021, 10, S88–S95. [Google Scholar] [CrossRef]
- Chafin, T.K.; Douglas, M.R.; Douglas, M.E. MrBait: Universal identification and design of targeted-enrichment capture probes. Bioinformatics 2018, 34, 4293–4296. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Duan, J.; Himsworth, C.; Hsiao, W.; Prystajecky, N.A. ProbeTools: Designing hybridization probes for targeted genomic sequencing of diverse and hypervariable viral taxa. BMC Genom. 2022, 23, 579. [Google Scholar] [CrossRef]
- Alanko, J.N.; Slizovskiy, I.B.; Lokshtanov, D.; Gagie, T.; Noyes, N.R.; Boucher, C. Syotti: Scalable bait design for DNA enrichment. Bioinformatics 2022, 38, i177–i184. [Google Scholar] [CrossRef]
- Faircloth, B.C. PHYLUCE Is a software package for the analysis of conserved genomic loci. Bioinformatics 2016, 32, 786–788. [Google Scholar] [CrossRef]
- Mayer, C.; Sann, M.; Donath, A.; Meixner, M.; Podsiadlowski, L.; Peters, R.S.; Petersen, M.; Meusemann, K.; Liere, K.; Wägele, J.-W.; et al. BaitFisher: A software package for multispecies target DNA enrichment probe design. Mol. Biol. Evol. 2016, 33, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.G. BaitsTools: Software for hybridization capture bait design. Mol. Ecol. Resour. 2018, 18, 356–361. [Google Scholar] [CrossRef]
- Jiménez-Mena, B.; Flávio, H.; Henriques, R.; Manuzzi, A.; Ramos, M.; Meldrup, D.; Edson, J.; Pálsson, S.; Ásta Ólafsdóttir, G.; Ovenden, J.R.; et al. Fishing for DNA? Designing baits for population genetics in target enrichment experiments: Guidelines, considerations and the new tool supeRbaits. Mol. Ecol. Resour. 2022, 22, 2105–2119. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Eckert, S.E.; Chan, J.Z.-M.; Houniet, D.; The PATHSEEK consortium; Breuer, J.; Speight, G. Enrichment by hybridisation of long DNA fragments for nanopore sequencing. Microb. Genom. 2016, 2, e000087. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.T.V.; Irinyi, L.; Hu, Y.; Schwessinger, B.; Meyer, W. Long-reads-based metagenomics in clinical diagnosis with a special focus on fungal infections. Front. Microbiol. 2022, 12, 708550. [Google Scholar] [CrossRef]
- Oehler, J.B.; Wright, H.; Stark, Z.; Mallett, A.J.; Schmitz, U. The application of long-read sequencing in clinical settings. Human. Genom. 2023, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Nicot, F.; Trémeaux, P.; Latour, J.; Carcenac, R.; Demmou, S.; Jeanne, N.; Ranger, N.; De Smet, C.; Raymond, S.; Dimeglio, C.; et al. Whole-genome single molecule real-time sequencing of SARS-CoV-2 Omicron. J. Med. Virol. 2023, 95, e28564. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quek, Z.B.R.; Ng, S.H. Hybrid-Capture Target Enrichment in Human Pathogens: Identification, Evolution, Biosurveillance, and Genomic Epidemiology. Pathogens 2024, 13, 275. https://doi.org/10.3390/pathogens13040275
Quek ZBR, Ng SH. Hybrid-Capture Target Enrichment in Human Pathogens: Identification, Evolution, Biosurveillance, and Genomic Epidemiology. Pathogens. 2024; 13(4):275. https://doi.org/10.3390/pathogens13040275
Chicago/Turabian StyleQuek, Z. B. Randolph, and Sock Hoon Ng. 2024. "Hybrid-Capture Target Enrichment in Human Pathogens: Identification, Evolution, Biosurveillance, and Genomic Epidemiology" Pathogens 13, no. 4: 275. https://doi.org/10.3390/pathogens13040275
APA StyleQuek, Z. B. R., & Ng, S. H. (2024). Hybrid-Capture Target Enrichment in Human Pathogens: Identification, Evolution, Biosurveillance, and Genomic Epidemiology. Pathogens, 13(4), 275. https://doi.org/10.3390/pathogens13040275