
Identification of Critical Amino Acid Residues of a Two-Component Sensor Protein for Signal Sensing in Porphyromonas gingivalis Fimbriation via Random Mutant Library Construction
 ,
,
Abstract
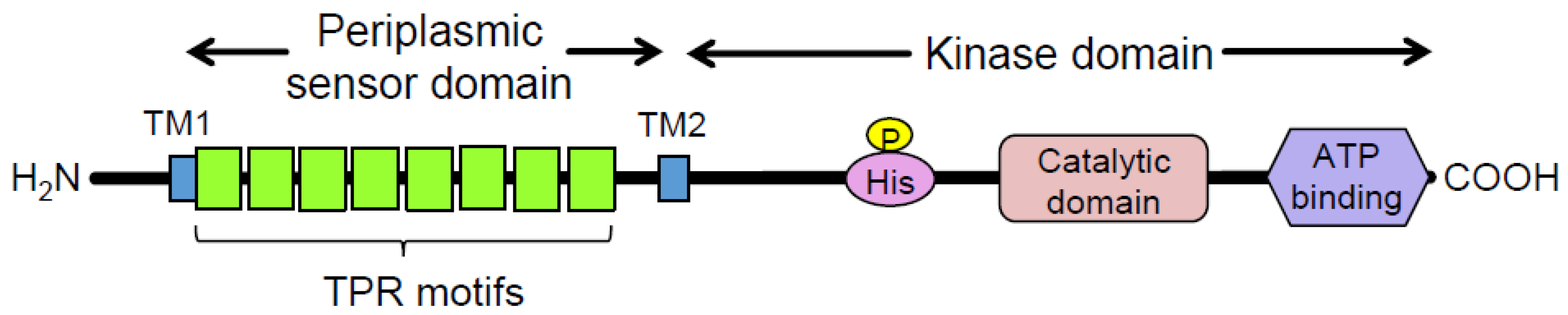
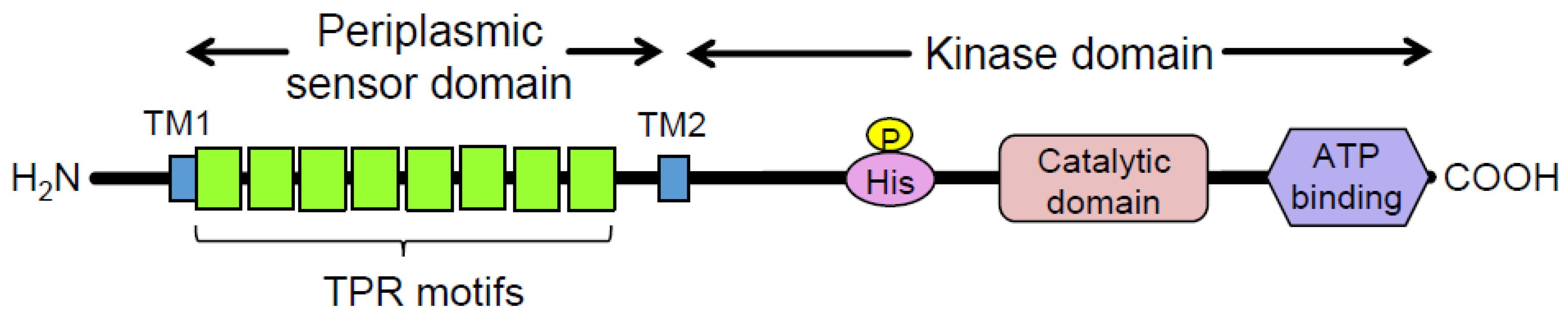
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
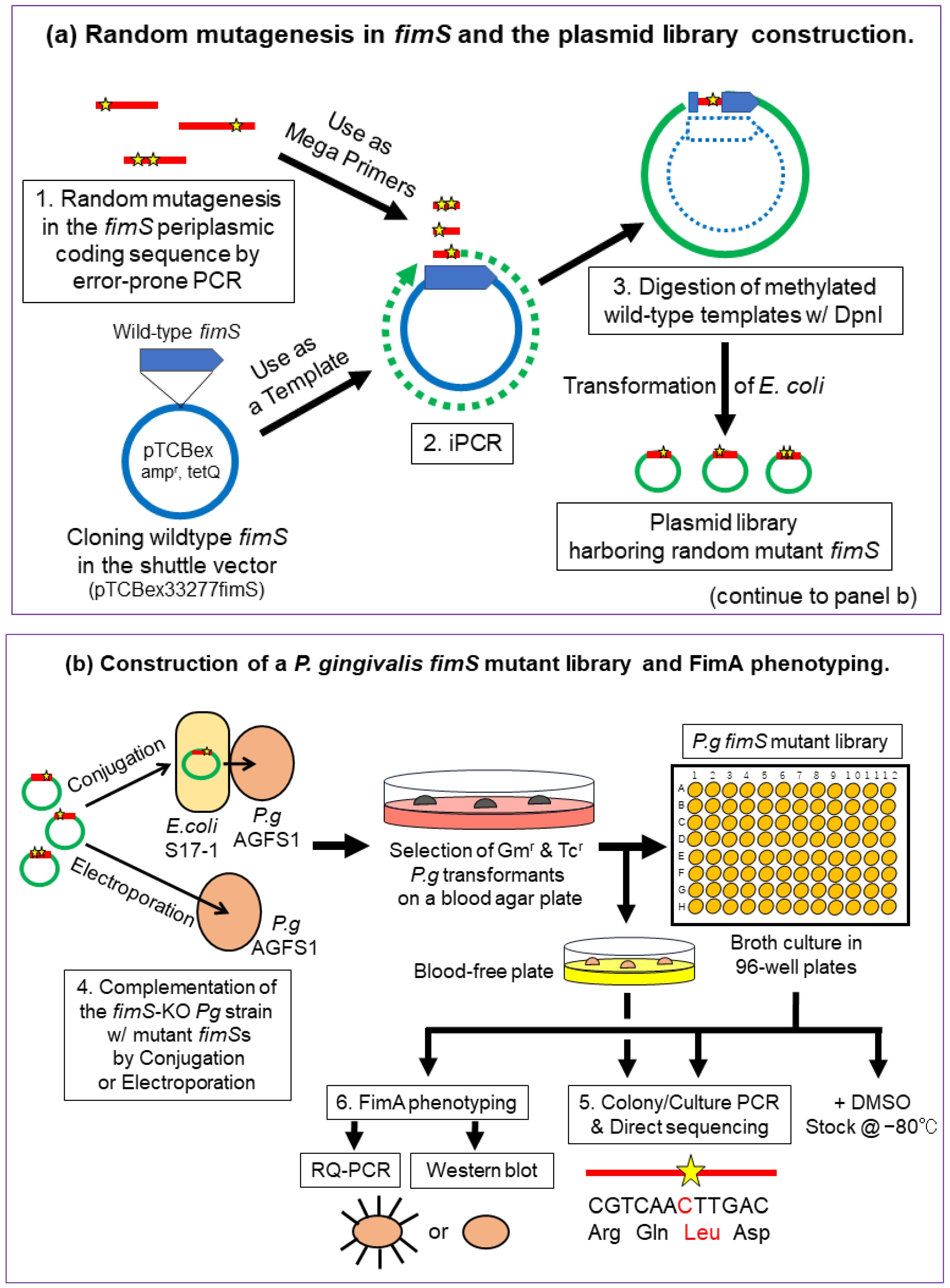
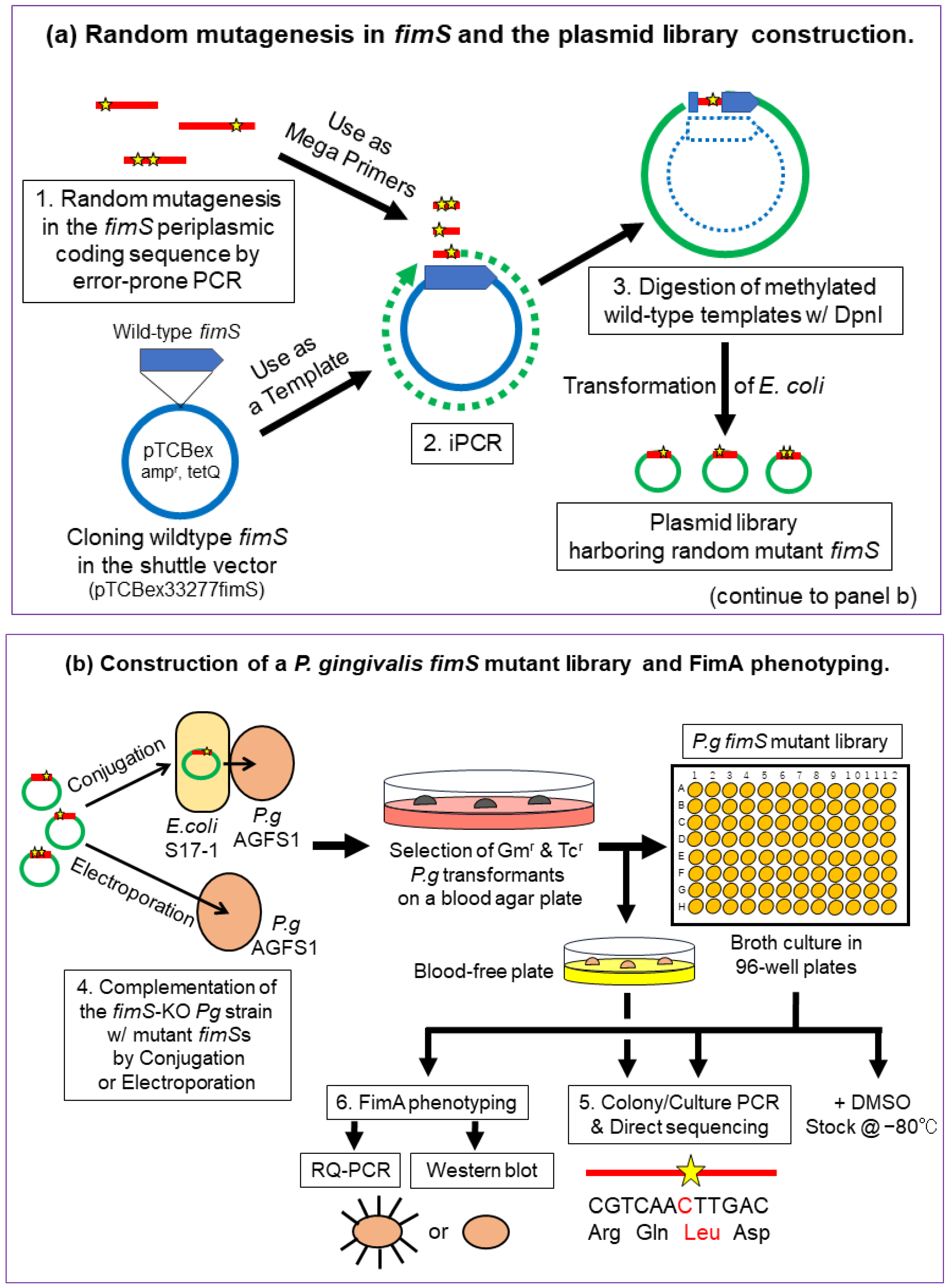
2.2. Overview of the Experimental Design
2.3. Construction of Pg Random Mutant Libraries
2.3.1. Error-Prone PCR
2.3.2. iPCR
2.3.3. Evaluation of the Mutation Rate
2.4. Introduction of the Mutant Plasmids into the fimS-KO Pg Strain by the E. coli–Pg Conjugal Transfer System or Electroporation
2.4.1. Electro-Transformation of E. coli S17-1 with iPCR Products
2.4.2. Conjugal Transfer of the Mutant Plasmid Library from S17-1 to fimS-KO Pg (AGFS1)
2.4.3. Introduction of the Mutant Plasmids into the fimS-KO Pg Strain by Electroporation
2.4.4. Subculture and Preparation of the Frozen Stocks of pTCBex-Introduced Pg Strains
2.5. Colony PCR and Direct Sequencing of the FimS Periplasmic-Domain-Coding Region
2.6. Preparation of RNA and RQ-PCR
2.7. Western Blot Analysis
3. Results
3.1. Evaluation of the Mutations Introduced by Error-Prone PCR
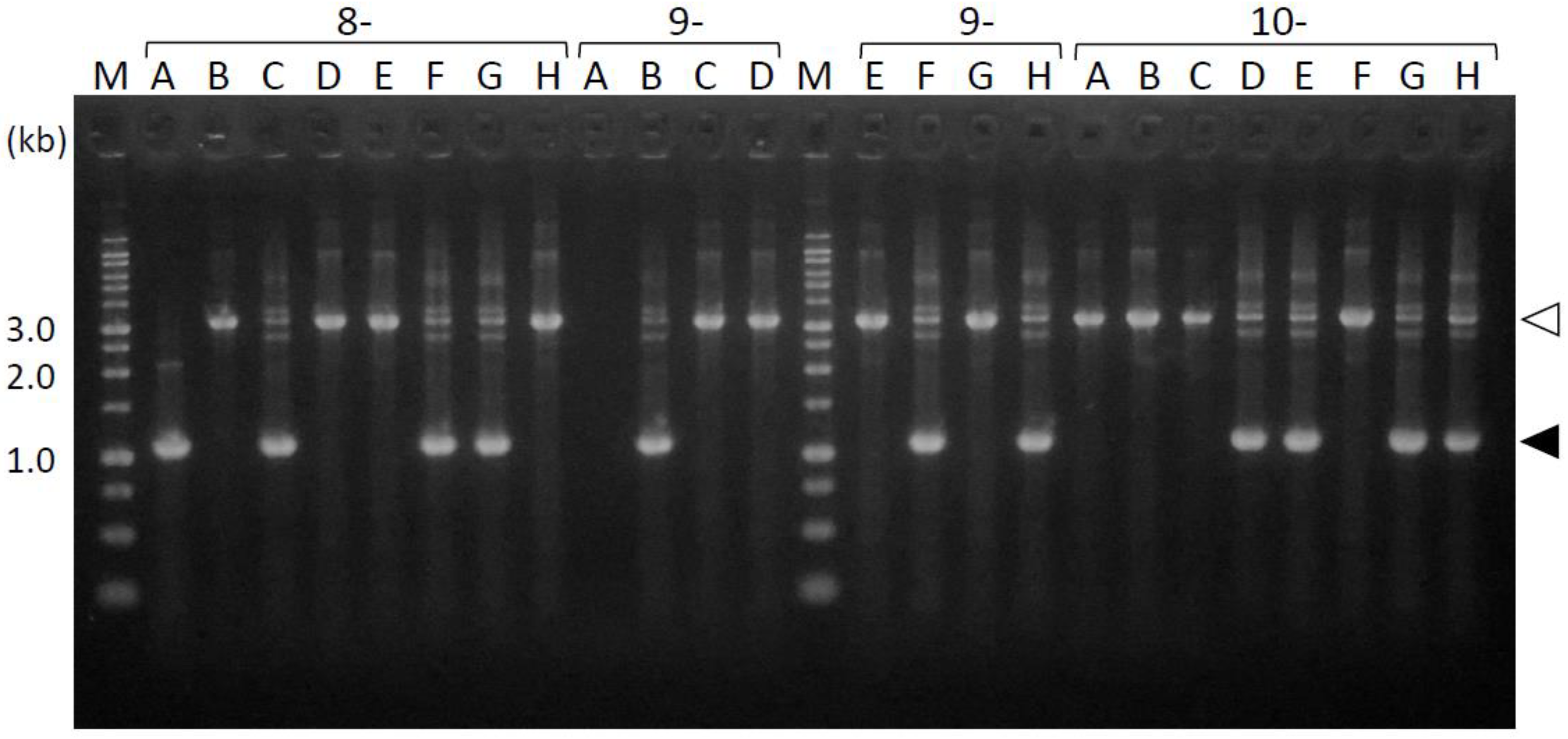
3.2. Comparison of the Transformation Efficiency of Pg between Conjugation and Electroporation
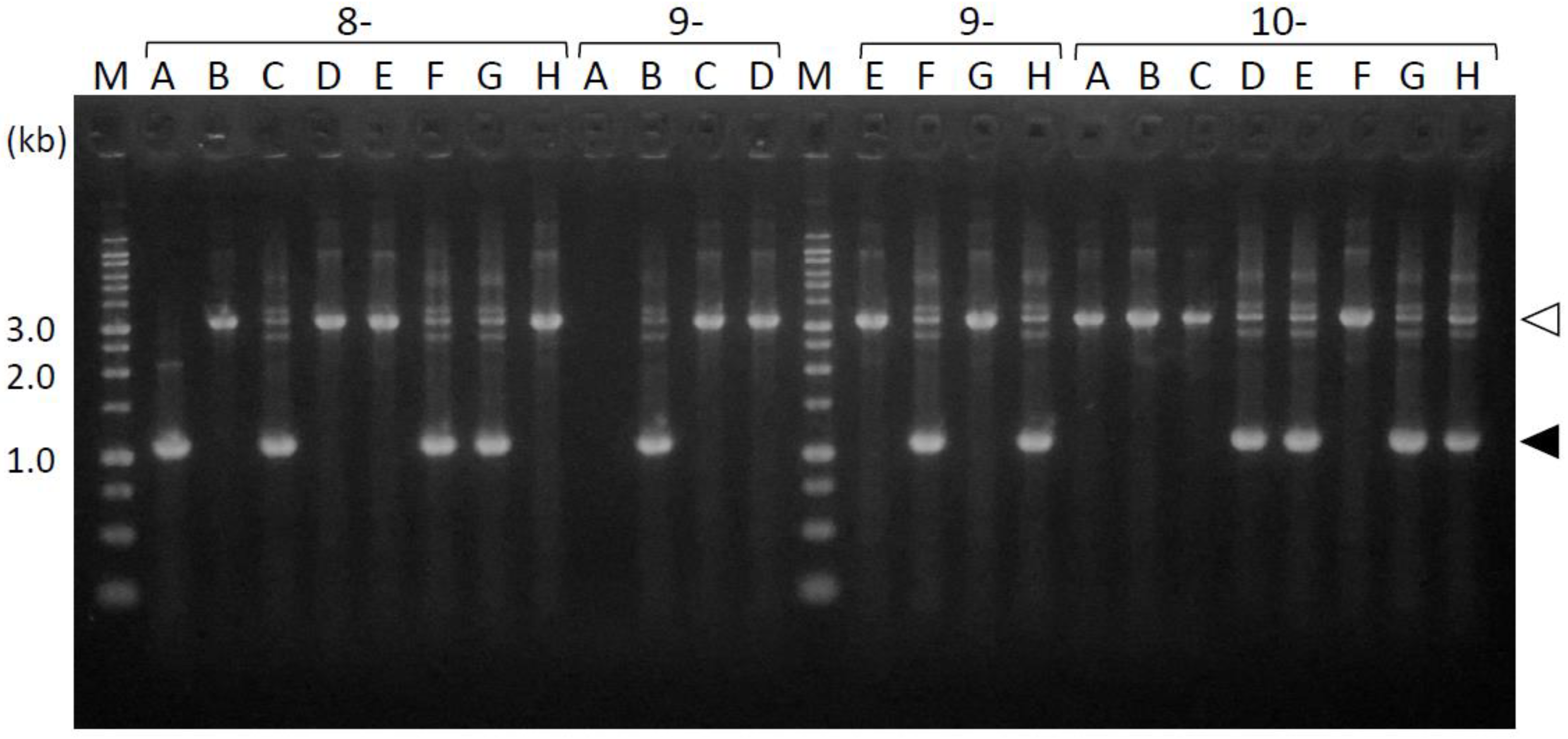
3.3. Amplification of the Periplasmic-Domain-Coding Sequences from the fimS-Complemented Pg Colonies
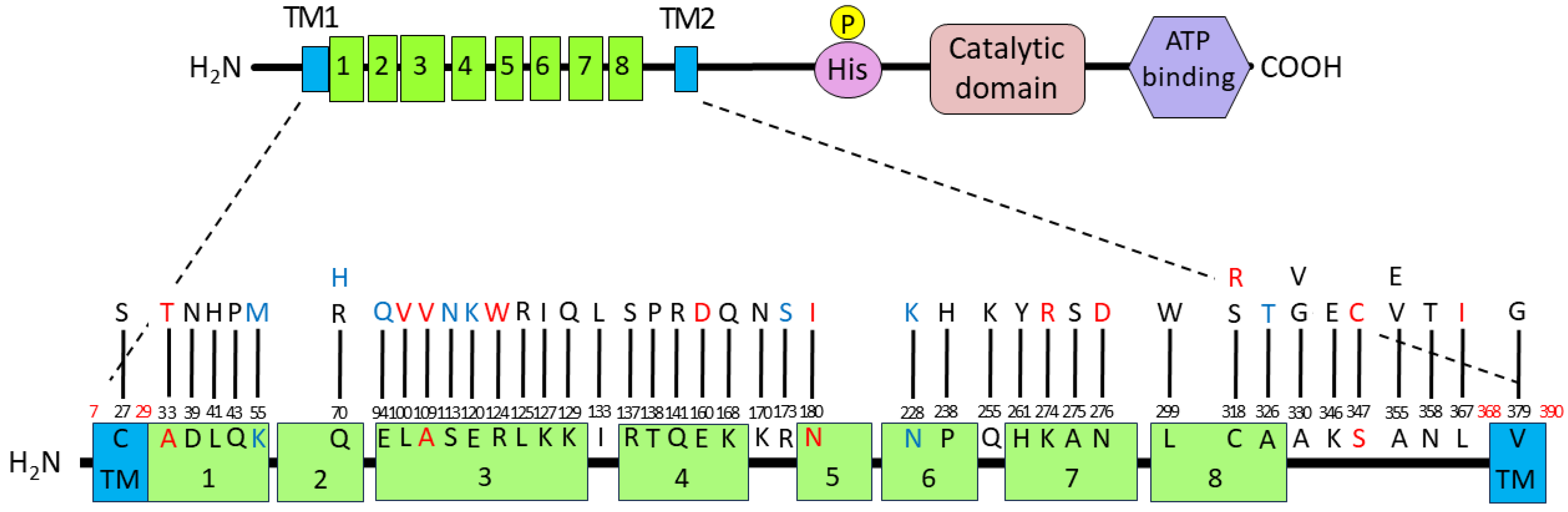
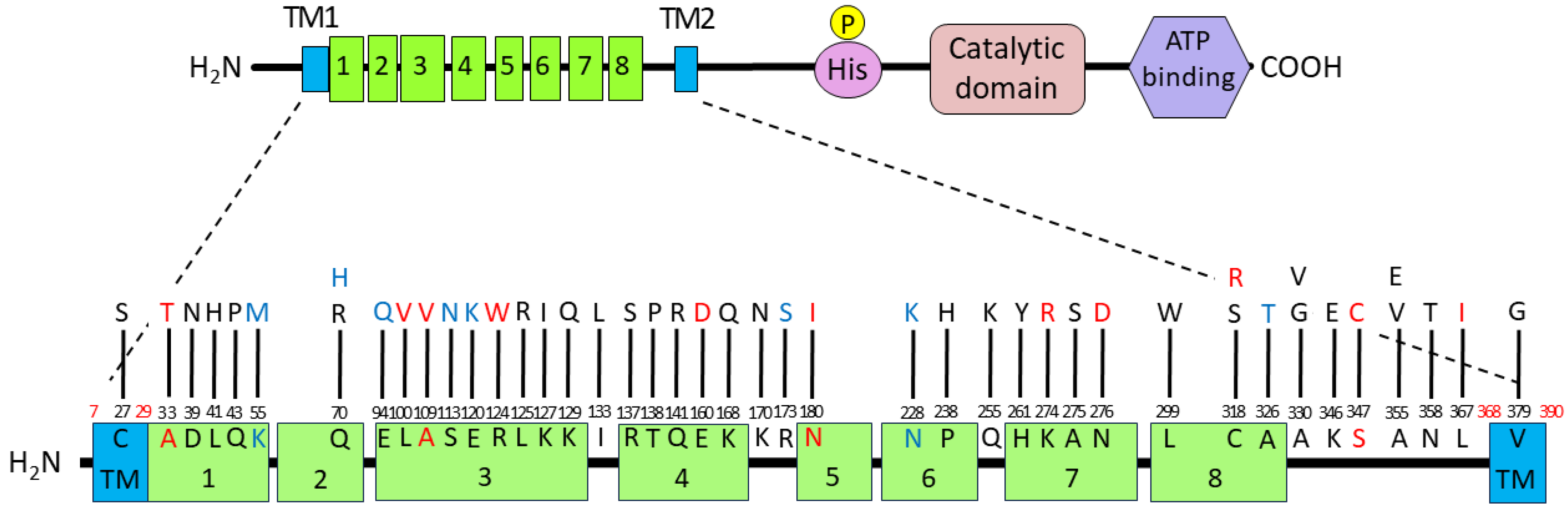
3.4. Introduced Mutations and Deduced Amino Acid Substitutions in the FimS Periplasmic Domain
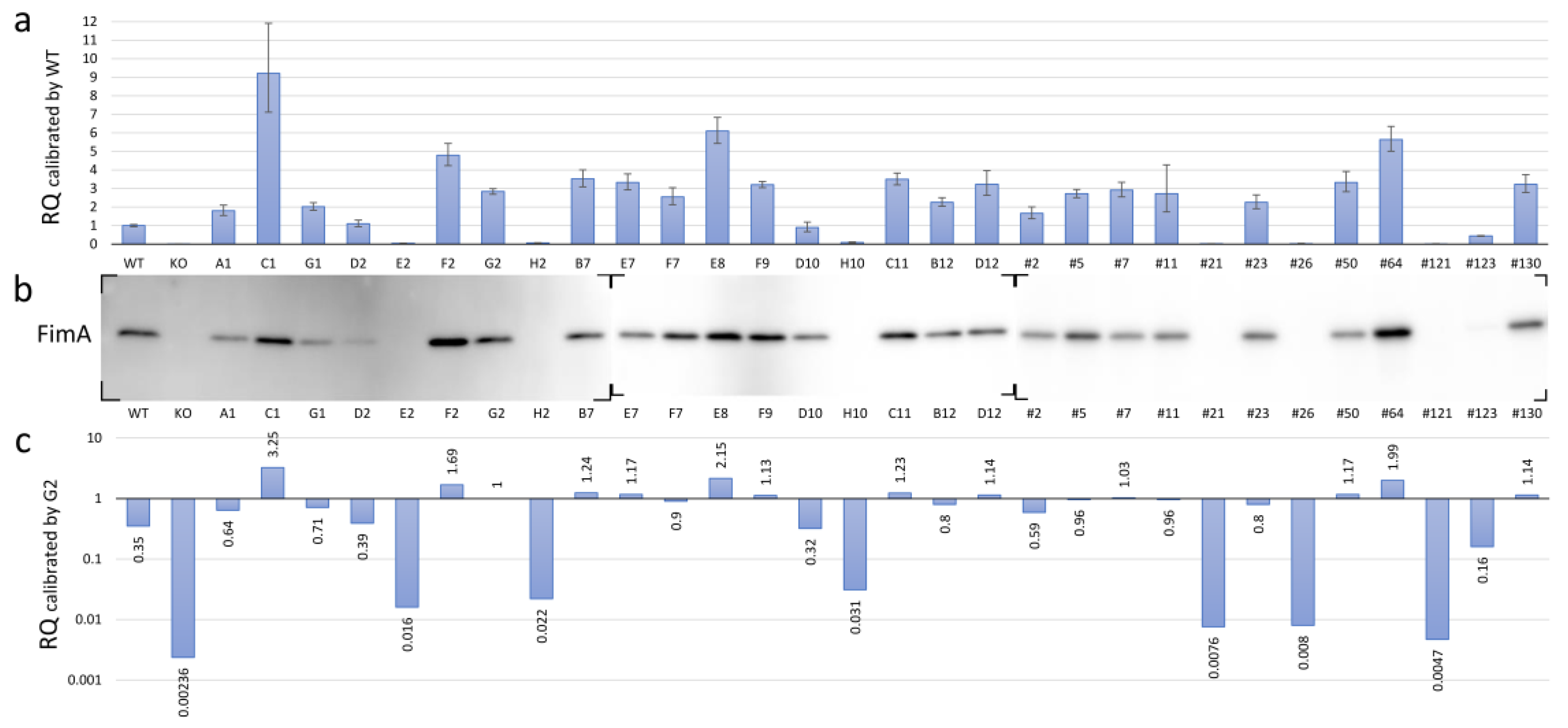
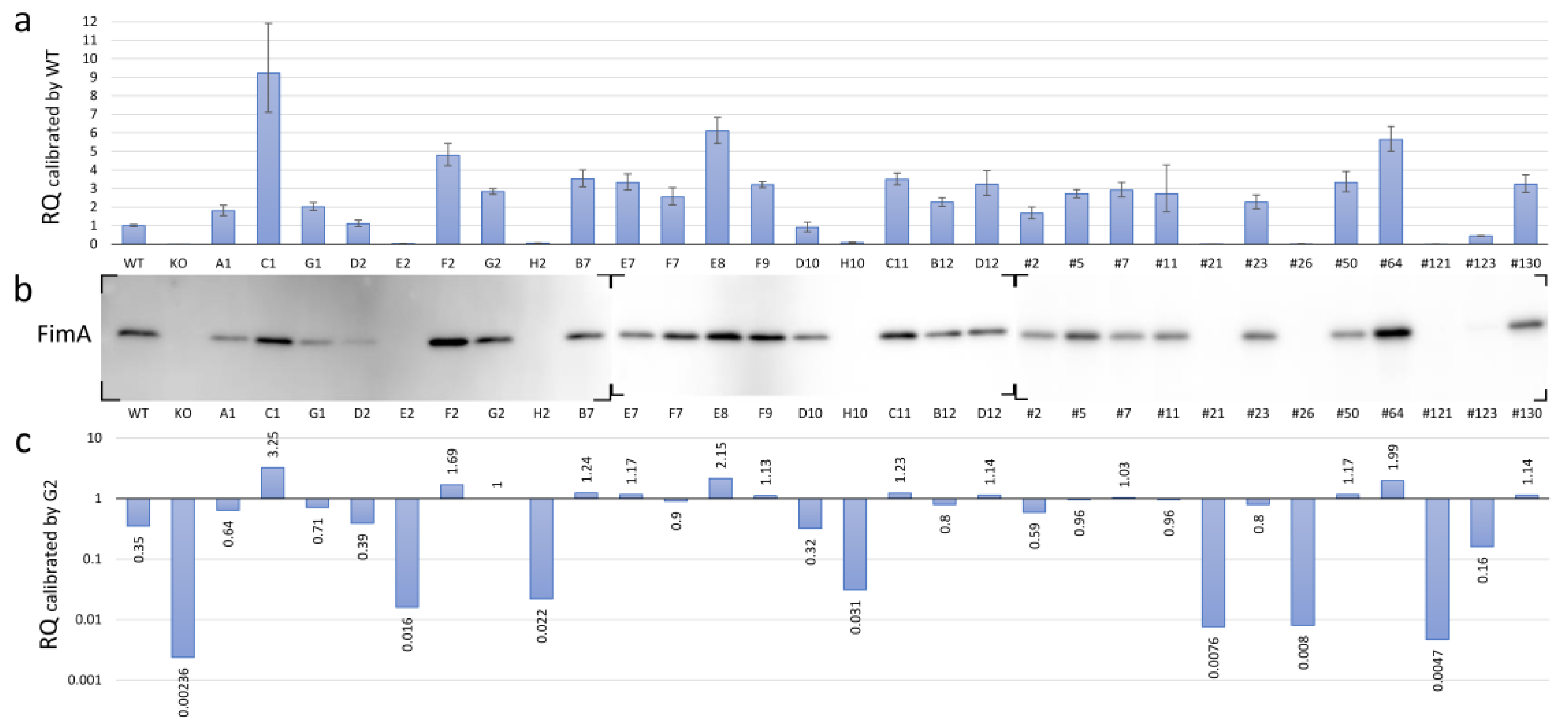
3.5. Expression of FimA at Transcriptional and Translational Levels in the fimS-Complemented AGFS1 Strain
3.6. Functional Mapping of the FimA Phenotypes Caused by Amino Acid Substitutions in the FimS Periplasmic Domain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Socransky, S.S.; Haffajee, A.D. Dental biofilms: Difficult therapeutic targets. Periodontol. 2000 2002, 28, 12–55. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T. The role of gingipains in the pathogenesis of periodontal disease. J. Periodontol. 2003, 74, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Potempa, J. Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J. Oral Microbiol. 2014, 6, 24800. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Wang, M.; Liang, S.; Triantafilou, M.; Triantafilou, K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc. Natl. Acad. Sci. USA 2008, 105, 13532–13537. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Yoshimura, F.; Duncan, M.J. A regulation cascade controls expression of Porphyromonas gingivalis fimbriae via the FimR response regulator. Mol. Microbiol. 2004, 54, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Duncan, M.J. Histidine kinase-mediated production and autoassembly of Porphyromonas gingivalis fimbriae. J. Bacteriol. 2010, 192, 1975–1987. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.M.; Robinson, V.L.; Goudreau, P.N. Two-component signal transduction. Annu. Rev. Biochem. 2000, 69, 183–215. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Yoshimura, F. The response regulator FimR is essential for fimbrial production of the oral anaerobe Porphyromonas gingivalis. Anaerobe 2001, 7, 255–262. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Karpenahalli, M.R.; Lupas, A.N.; Söding, J. TPRpred: A tool for prediction of TPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinform. 2007, 8, 2. [Google Scholar] [CrossRef]
- Cerveny, L.; Straskova, A.; Dankova, V.; Hartlova, A.; Ceckova, M.; Staud, F.; Stulik, J. Tetratricopeptide repeat motifs in the world of bacterial pathogens: Role in virulence mechanisms. Infect. Immun. 2013, 81, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Kawamura, A.; Nishikawa, K.; Iida, H.; Miyazawa, K.; Goto, S.; Hasegawa, Y. The AtoC family response regulator upregulates an operon encoding putative outer membrane proteins sorted by type IX secretion system in Porphyromonas gingivalis. J. Oral Biosci. 2023, 65, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K. Mutational analysis of the periplasmic sensor domain of the Porphyromonas gingivalis FimS histidine kinase. In Microbe; American Society for Microbiology: Boston, MA, USA, 2016; abstract 757. [Google Scholar]
- Fujii, K.; Minagawa, H.; Terada, Y.; Takaha, T.; Kuriki, T.; Shimada, J.; Kaneko, H. Use of random and saturation mutageneses to improve the properties of Thermus aquaticus amylomaltase for efficient production of cycloamyloses. Appl. Environ. Microbiol. 2005, 71, 5823–5827. [Google Scholar] [CrossRef] [PubMed]
- Mordukhova, E.A.; Lee, H.S.; Pan, J.G. Improved thermostability and acetic acid tolerance of Escherichia coli via directed evolution of homoserine o-succinyltransferase. Appl. Environ. Microbiol. 2008, 74, 7660–7668. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K. Creating random mutagenesis libraries by megaprimer PCR of whole plasmid (MEGAWHOP). Methods Mol. Biol. 2003, 231, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, R.C.; Joyce, G.F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992, 2, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Blatch, G.L.; Lässle, M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. BioEssays 1999, 21, 932–939. [Google Scholar] [CrossRef]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Dain Md Opo, F.A.; Alsaiari, A.A.; Rahman Molla, M.H.; Ahmed Sumon, M.A.; Yaghmour, K.A.; Ahammad, F.; Mohammad, F.; Simal-Gandara, J. Identification of novel natural drug candidates against BRAF mutated carcinoma; An integrative in-silico structure-based pharmacophore modeling and virtual screening process. Front. Chem. 2022, 10, 986376. [Google Scholar] [CrossRef] [PubMed]
- Lin-Goerke, J.L.; Robbins, D.J.; Burczak, J.D. PCR-Based Random Mutagenesis Using Manganese and Reduced dNTP Concentration. BioTechniques 1997, 23, 409–412. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain, Plasmid, or Primer | Characteristics 1 or Sequences (5′-3′; Amplicon Size) | Source or Reference |
|---|---|---|
| Porphyromonas gingivalis ATCC 33277 AGFS1 Escherichia coli DH5α BL21-Gold S17-1 Plasmids pTCBex pTCBex33277fimS pTCBexdm Primers 2 error-prone/colony PCR FimSperi5/3 direct sequencing FIMSSFW3.5 FIMSSFW4rv RQ-PCR Pg16S rRNA-F/R 33277 fimA-F/R | parent type strain (WT) 33277 with an ermF-AM cassete insertion in fimS, Emr a host for general/PCR cloning a host for plasmid construction/cloning a host for conjugal transfer of pTCBex to P. gingivalis, Tp r a P. gingivalis expression vector, Apr, Tc r a pTCBex carrying 33277 fimS ORF Mob region-deleted pTCBex ACGCGACTAACTATCCTGACATTTCTTGGA/ ATGGTTGCCACCAACAGTGTGGATATTATG; 1097 bp ATAGCCTACAACAACATGGCTAAC CATGAGACTGTTGCACATACTTACA GTCAATGGGCGAGAGCCTGAA/ AGTGTCAGTCGCAGTATGGCAA; 383 bp CTTGTAACAAAGACAACGAGGCAG/ GCAGGTGCAACGTAATTACGGCTC; 700 bp | ATCC Nishikawa and Duncan [6] Invitrogen Agilent Simon et al. [12] Nishikawa and Duncan [6], Kawamura et al. [13] Nishikawa and Duncan [6] Nishikawa [14] This study This study This study This study Nishikawa and Duncan [6] Nishikawa and Duncan [6] |
| Strain/Clone 1 | Mutation(s) in fimS Gene |
Deduced Amino Acid Substitution(s)
in FimS Periplasmic Domain | Accession No. |
|---|---|---|---|
| A1 | G115A, A209G, T896G, T1136G | D39N, Q70R, L299W, V379G in TM2 | LC800510 |
| C1 | C189A, A210C, G280C, T330G, G338A, G358A | Q70H, E94Q, S113N, E120K | LC800511 |
| G1 | A502C | K168Q | LC800512 |
| D2 | A480T, T952C, A1073C | E160D, C318R, N358T | LC800513 |
| E2 | A370C, C429T, A480T, A821G, A826G, T952C | E160D, K274R, N276D, C318R | LC800514 |
| F2 | A164T | K55M | LC800515 |
| H2 | A1039T | S347C | LC800516 |
| B7 | A1073C | N358T | LC800517 |
| E7 | T122A | L41H | LC800518 |
| F7 | C1064T | A355V | LC800519 |
| E8 | T684A | N228K | LC800520 |
| F9 | C409A | R137S | LC800521 |
| D10 | C713A, G823T | P238H, A275S | LC800522 |
| H10 | T298G, A370T | L100V, R124W | LC800523 |
| C11 | C763A | Q255K | LC800524 |
| B12 | C989T | A330V | LC800525 |
| D12 | T896G | L299W | LC800526 |
| #2 | G510T, A1036G | K170N, K346E | LC800527 |
| #5 | A385C, C1064A | K129Q, A355E | LC800528 |
| #7 | C781T, C989G | H261Y, A330G | LC800529 |
| #11 | A128C, T333C, A380T, A397C, A412C, A422G | Q43P, K127I, I133L, T138P, Q141R | LC800530 |
| #21 | A539T | N180I | LC800531 |
| #23 | C713A | P238H | LC800532 |
| #26 | C326T, T1099A | A109V, L367I | LC800533 |
| #50 | T79A | C27S in TM1 | LC800534 |
| #64 | A519T, G976A | R173S, A326T | LC800535 |
| #121 | C326T | A109V | LC800536 |
| #123 | G97A | A33T | LC800537 |
| #130 | T374G, T952A | L125R, C318S | LC800538 |
| Sample 1 | AvgΔCt 2 | ΔCt SD | Calibrated by WT | Calibrated by G2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔΔCt | RQ | RQ Min | RQ Max | ΔΔCt | RQ | RQ Min | RQ Max | |||
| WT KO G2 A1 C1 G1 D2 E2 F2 H2 B7 E7 F7 E8 F9 D10 H10 C11 B12 D12 #2 #5 #7 #11 #21 #23 #26 #50 #64 #121 #123 #130 | 4.604 21.827 13.101 13.757 11.402 13.594 14.467 19.050 12.343 18.617 12.789 12.870 13.255 11.997 12.923 14.768 18.122 12.799 13.430 12.914 13.875 13.162 13.060 13.159 20.137 13.431 20.075 12.870 12.109 20.837 15.784 12.912 | 0.030 0.047 0.022 0.049 0.081 0.032 0.050 0.053 0.039 0.056 0.041 0.041 0.056 0.036 0.017 0.089 0.089 0.029 0.032 0.064 0.059 0.026 0.042 0.141 0.044 0.052 0.162 0.051 0.037 0.099 0.017 0.046 | 0 7.222 −1.504 −0.848 −3.203 −1.011 −0.138 4.446 −2.261 4.012 −1.816 −1.735 −1.350 −2.608 −1.681 0.163 3.518 −1.806 −1.174 −1.691 −0.729 −1.442 −1.544 −1.446 5.533 −1.173 5.470 −1.734 −2.496 6.232 1.179 −1.692 | 1 0.007 2.836 1.8 9.208 2.015 1.100 0.0459 4.795 0.062 3.521 3.328 2.549 6.095 3.207 0.893 0.087 3.496 2.257 3.229 1.658 2.718 2.916 2.724 0.022 2.256 0.023 3.327 5.641 0.013 0.442 3.232 | 0.941 0.006 2.691 1.538 7.114 1.821 0.938 0.0387 4.236 0.0519 3.092 2.921 2.131 5.434 3.042 0.673 0.066 3.192 2.035 2.630 1.372 2.503 2.548 1.735 0.019 1.912 0.013 2.825 5.009 0.010 0.418 2.788 | 1.062 0.007 2.989 2.107 11.918 2.229 1.290 0.054 5.427 0.074 4.010 3.792 3.048 6.836 3.381 1.185 0.116 3.829 2.503 3.963 2.003 2.950 3.338 4.278 0.025 2.660 0.038 3.918 6.353 0.018 0.467 3.746 | 1.504 8.726 0 0.656 −1.699 0.493 1.366 5.950 −0.757 5.516 −0.312 −0.231 0.154 −1.104 −0.177 1.667 5.022 −0.302 0.330 −0.187 0.775 0.062 −0.040 0.058 7.037 0.331 6.974 −0.230 −0.992 7.736 2.683 −0.188 | 0.353 0.002 1 0.635 3.247 0.710 0.388 0.016 1.690 0.0219 1.241 1.173 0.899 2.149 1.131 0.315 0.031 1.233 0.796 1.138 0.584 0.958 1.028 0.960 0.008 0.795 0.008 1.173 1.989 0.005 0.156 1.139 | 0.332 0.002 0.949 0.542 2.508 0.642 0.331 0.014 1.493 0.0183 1.090 1.030 0.751 1.916 1.073 0.237 0.023 1.125 0.718 0.927 0.484 0.883 0.898 0.612 0.007 0.674 0.005 0.996 1.766 0.003 0.147 0.983 | 0.375 0.003 1.054 0.743 4.202 0.786 0.455 0.019 1.914 0.026 1.414 1.337 1.075 2.410 1.192 0.418 0.041 1.350 0.882 1.397 0.706 1.040 1.177 1.508 0.009 0.938 0.013 1.381 2.240 0.006 0.165 1.321 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iida, H.; Nishikawa, K.; Sato, T.; Kawaguchi, M.; Miyazawa, K.; Hasegawa, Y. Identification of Critical Amino Acid Residues of a Two-Component Sensor Protein for Signal Sensing in Porphyromonas gingivalis Fimbriation via Random Mutant Library Construction. Pathogens 2024, 13, 309. https://doi.org/10.3390/pathogens13040309
Iida H, Nishikawa K, Sato T, Kawaguchi M, Miyazawa K, Hasegawa Y. Identification of Critical Amino Acid Residues of a Two-Component Sensor Protein for Signal Sensing in Porphyromonas gingivalis Fimbriation via Random Mutant Library Construction. Pathogens. 2024; 13(4):309. https://doi.org/10.3390/pathogens13040309
Chicago/Turabian StyleIida, Haruka, Kiyoshi Nishikawa, Takuma Sato, Misuzu Kawaguchi, Ken Miyazawa, and Yoshiaki Hasegawa. 2024. "Identification of Critical Amino Acid Residues of a Two-Component Sensor Protein for Signal Sensing in Porphyromonas gingivalis Fimbriation via Random Mutant Library Construction" Pathogens 13, no. 4: 309. https://doi.org/10.3390/pathogens13040309