

Novel Betanucleorhabdoviruses Infecting Elderberry (Sambucus nigra L.): Genome Characterization and Genetic Variability

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Isolation
2.3. High-Throughput Sequencing
2.4. High-Throughput Sequencing Data Analysis
2.5. Completing Virus Sequence, RACE-PCR and Sanger Sequencing
2.6. RT-PCR Virus Detection
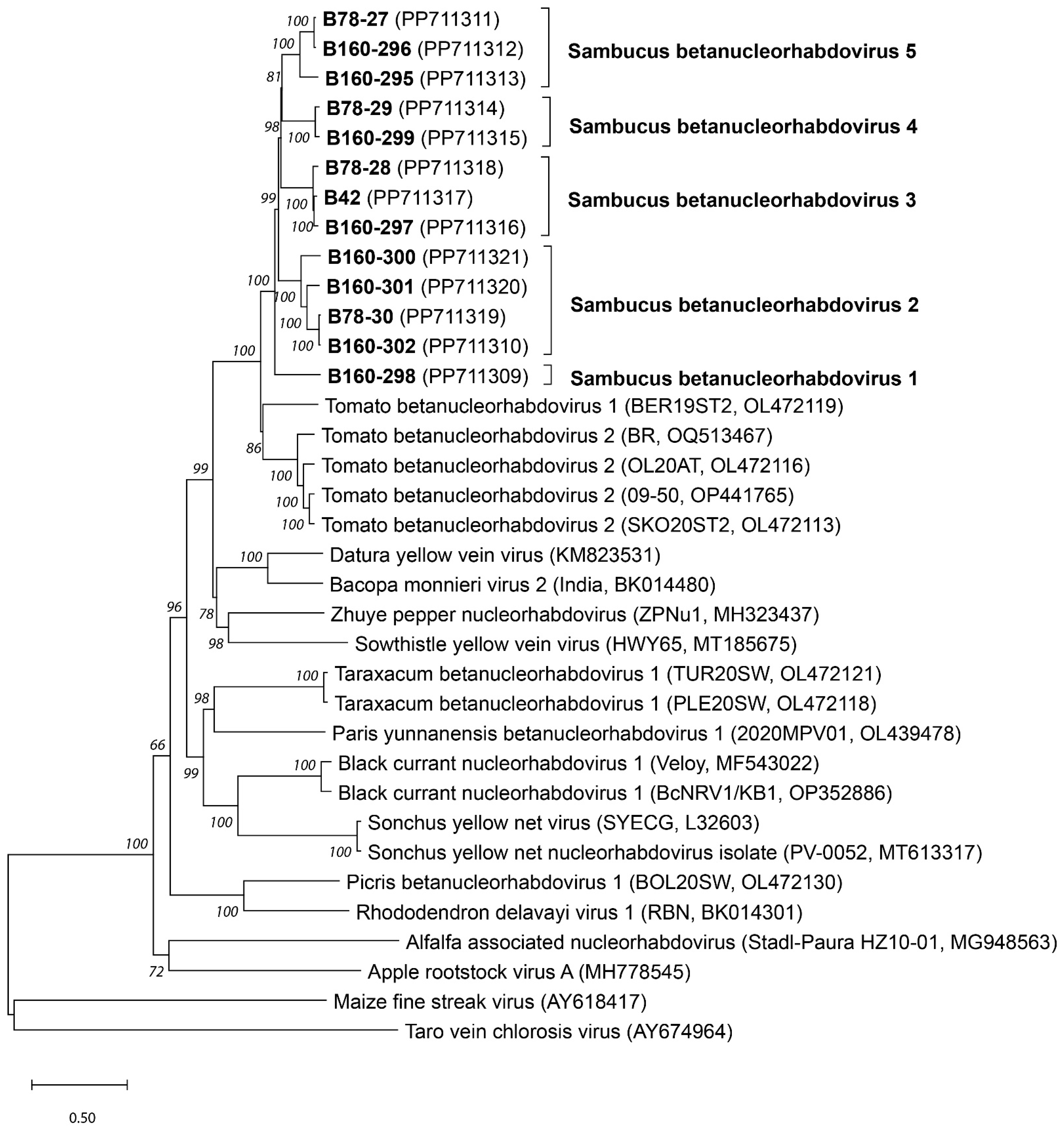
2.7. Genetic and Phylogenetic Analyses
2.8. Biological Testing–Transmission Trials
3. Results
3.1. Genome Organization of the Sambucus Betanucleorhabdoviruses
3.1.1. Sambucus Betanucleorhabdovirus 1
3.1.2. Sambucus Betanucleorhabdovirus 2
3.1.3. Sambucus Betanucleorhabdovirus 3
3.1.4. Sambucus Betanucleorhabdovirus 4
3.1.5. Sambucus Betanucleorhabdovirus 5
3.2. Genome Analysis of Sambucus Betanucleorhabdoviruses
3.3. Preliminary Results on Sambucus Betanucleorhabdoviruses Spread Mechanisms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bejerman, N.; Dietzgen, R.G.; Debat, H. Illuminating the plant rhabdovirus landscape through metatranscriptomics data. Viruses 2021, 13, 1304. [Google Scholar] [CrossRef]
- Walker, P.J.; Freitas-Astúa, J.; Bejerman, N.; Blasdell, K.R.; Breyta, R.; Dietzgen, R.G.; Fooks, A.R.; Kondo, H.; Kurath, G.; Kuzmin, I.V.; et al. ICTV Virus Taxonomy Profile: 2022. J. Gen. Virol. 2022, 103, 001689. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avsic-Zupanc, T.; Ayllon, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2020 taxonomic update for phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 165, 3023–3072. [Google Scholar] [CrossRef]
- Jackson, A.O.; Dietzgen, R.G.; Goodin, M.M.; Bragg, J.N.; Deng, M. Biology of plant rhabdoviruses. Annu. Rev. Phytopathol. 2005, 43, 623–660. [Google Scholar] [CrossRef] [PubMed]
- Belete, M.T.; Igori, D.; Kim, S.E.; Lee, S.H.; Moon, J.S. Complete genome sequence of cnidium virus 1, a novel betanucleorhabdovirus infecting Cnidium officinale. Arch. Virol. 2022, 167, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Abe, J.; Adkins, S.; Alkhovsky, S.V.; Avsic-Zupanc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; Ballinger, M.J.; Baranwal, V.K.; et al. Annual (2023) taxonomic update of RNA-directed RNA polymerase-encoding negative-sense RNA viruses (realm: Kingdom: Phylum). J. Gen. Virol. 2023, 104, 001864. [Google Scholar] [CrossRef] [PubMed]
- Sidharthan, V.K.; Baranwal, V.K. Mining of the water hyssop (Bacopa monnieri) transcriptome reveals genome sequences of two putative novel rhabdoviruses and a solendovirus. Arch. Virol. 2021, 166, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.I.; Pamitha, N.S.; Naveen, K.P.; Biju, C.N. Identification and characterization of cardamom vein clearing virus, a novel aphid-transmitted nucleorhabdovirus. Eur. J. Plant Pathol. 2020, 156, 1053–1062. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Innes, D.J.; Bejerman, N. Complete genome sequence and intracellular protein localization of Datura yellow vein nucleorhabdovirus. Virus Res. 2015, 205, 7–11. [Google Scholar] [CrossRef]
- Debat, H.J.; Bejerman, N. Novel bird’s-foot trefoil RNA viruses provide insights into a clade of legume-associated enamoviruses and rhabdoviruses. Arch. Virol. 2019, 164, 1419–1426. [Google Scholar] [CrossRef]
- Baek, D.; Lim, S.; Ju, H.J.; Kim, H.R.; Lee, S.H.; Moon, J.S. The complete genome sequence of apple rootstock virus A, a novel nucleorhabdovirus identified in apple rootstocks. Arch. Virol. 2019, 164, 2641–2644. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Richert-Poggeler, K.R.; Maass, C.; Vetten, H.J.; Ziebell, H. Characterisation of a novel nucleorhabdovirus infecting alfalfa (Medicago sativa). Virol. J. 2019, 16, 55. [Google Scholar] [CrossRef] [PubMed]
- Rivarez, M.P.S.; Pecman, A.; Bacnik, K.; Maksimovic, O.; Vucurovic, A.; Seljak, G.; Mehle, N.; Gutiérrez-Aguirre, I.; Ravnikar, M.; Kutnjak, D. In-depth study of tomato and weed viromes reveals undiscovered plant virus diversity in an agroecosystem. Microbiome 2023, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Heaton, L.A.; Hillman, B.I.; Hunter, B.G.; Zuidema, D.; Jackson, A.O. Physical map of the genome of sonchus yellow net virus, a plant rhabdovirus with 6 genes and conserved gene junction sequences. Proc. Natl. Acad. Sci. USA 1989, 86, 8665–8668. [Google Scholar] [CrossRef]
- Wu, L.P.; Yang, T.; Liu, H.W.; Postman, J.; Li, R. Molecular characterization of a novel rhabdovirus infecting blackcurrant identified by high-throughput sequencing. Arch. Virol. 2018, 163, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Stenger, D.C.; Burbank, L.P.; Wang, R.Y.; Stewart, A.A.; Mathias, C.; Goodin, M.M. Lost and found: Rediscovery and genomic characterization of sowthistle yellow vein virus after a 30+year hiatus. Virus Res. 2020, 284, 197987. [Google Scholar] [CrossRef]
- Cao, M.J.; Zhang, S.; Li, M.; Liu, Y.J.; Dong, P.; Li, S.R.; Kuang, M.; Li, R.H.; Zhou, Y. Discovery of four novel viruses associated with flower yellowing disease of Green sichuan pepper (Zanthoxylum armatum) by virome analysis. Viruses 2019, 11, 696. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). EC 55, Jena, Germany, August 2023. Email Ratification April 2024. 2024. Available online: https://ictv.global/msl (accessed on 15 May 2024).
- Hu, J.Y.; Miao, T.L.; Que, K.J.; Rahman, M.S.; Zhang, L.; Dong, X.; Ji, P.Z.; Dong, J.H. Identification, molecular characterization and phylogenetic analysis of a novel nucleorhabdovirus infecting Paris polyphylla var. yunnanensis. Sci. Rep. 2023, 13, 10040. [Google Scholar] [CrossRef]
- Atkinson, M.D.; Atkinson, E. Sambucus nigra L. J. Ecol. 2002, 90, 895–923. [Google Scholar] [CrossRef]
- Plants of the World Online. Royal Botanic Gardens, Kew. Available online: https://powo.science.kew.org/ (accessed on 16 April 2024).
- Ferreira, S.S.; Martins-Gomes, C.; Nunes, F.M.; Silva, A.M. Elderberry (Sambucus nigra L.) extracts promote anti-inflammatory and cellular antioxidant activity. Food Chem. X 2022, 15, 100437. [Google Scholar] [CrossRef]
- Mocanu, M.L.; Amariei, S. Elderberries-A source of bioactive compounds with antiviral action. Plants 2022, 11, 740. [Google Scholar] [CrossRef]
- Dominguez, R.; Pateiro, M.; Munekata, P.E.S.; Santos Lopez, E.M.; Rodriguez, J.A.; Barros, L.; Lorenzo, J.M. Potential use of elderberry (Sambucus nigra L.) as natural colorant and antioxidant in the food industry. A Review. Foods 2021, 10, 2713. [Google Scholar] [CrossRef]
- Dominguez, R.; Zhang, L.; Rocchetti, G.; Lucini, L.; Pateiro, M.; Munekata, P.E.S.; Lorenzo, J.M. Elderberry (Sambucus nigra L.) as potential source of antioxidants. Characterization, optimization of extraction parameters and bioactive properties. Food Chem. 2020, 330, 127266. [Google Scholar] [CrossRef] [PubMed]
- Festa, J.; Hussain, A.; Hackney, A.; Desai, U.; Sahota, T.S.; Singh, H.; Da Boit, M. Elderberry extract improves molecular markers of endothelial dysfunction linked to atherosclerosis. Food Sci. Nutr. 2023, 11, 4047–4059. [Google Scholar] [CrossRef]
- Liu, D.; He, X.Q.; Wu, D.T.; Li, H.B.; Feng, Y.B.; Zou, L.; Gan, R.Y. Elderberry (Sambucus nigra L.): Bioactive compounds, health functions, and applications. J. Agric. Food Chem. 2022, 70, 4202–4220. [Google Scholar] [CrossRef] [PubMed]
- Šafářová, D.; Vavroušková, K.; Candresse, T.; Navrátil, M. Molecular characterization of a novel Aureusvirus infecting elderberry (Sambucus nigra L.). PLoS ONE 2018, 13, e0200506. [Google Scholar] [CrossRef] [PubMed]
- Gentit, P.; Foissac, X.; Svanella-Dumas, L.; Peypelut, M.; Candresse, T. Characterization of two different apricot latent virus variants associated with peach asteroid spot and peach sooty ringspot diseases. Arch. Virol. 2001, 146, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11 Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Ba, A.N.; Pogoutse, A.; Provart, N.; Moses, A.M. NLStradamus: A simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinform. 2009, 10, 202. [Google Scholar] [CrossRef]
- Šafářová, D.; Candresse, T.; Navrátil, M. Complete genome sequence of a novel cytorhabdovirus infecting elderberry (Sambucus nigra L.) in the Czech Republic. Arch. Virol. 2022, 167, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Šafářová, D.; Candresse, T.; Navrátil, M. Complete genome sequence of a novel bromovirus infecting elderberry (Sambucus nigra L.) in the Czech Republic. Arch. Virol. 2018, 163, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Mlynarczyk, K.; Walkowiak-Tomczak, D.; Lysiak, G.P. Bioactive properties of Sambucus nigra L. as a functional ingredient for food and pharmaceutical industry. J. Funct. Foods 2018, 40, 377–390. [Google Scholar] [CrossRef]
- Ho, T.; Quito-Avila, D.; Keller, K.E.; Postman, J.D.; Martin, R.R.; Tzanetakis, I.E. Evidence of sympatric speciation of elderberry carlaviruses. Virus Res. 2016, 215, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Vainio, E.J.; Rumbou, A.; Diez, J.J.; Büttner, C. Forest tree virome as a source of tree diseases and biological control agents. Curr. For. Rep. 2024, 10, 153–174. [Google Scholar] [CrossRef]
- Maliogka, V.I.; Minafra, A.; Saldarelli, P.; Ruiz-Garcia, A.B.; Glasa, M.; Katis, N.; Olmos, A. Recent advances on detection and characterization of fruit tree viruses using high-throughput sequencing technologies. Viruses 2018, 10, 436. [Google Scholar] [CrossRef]
- ICTV Taxonomy Release. 2024. Available online: https://ictv.global/taxonomy (accessed on 27 April 2024).
- Thomas, J.E.; Dietzgen, R.G. Characterization of Datura yellow vein virus, a newly described rhabdovirus from Australia. Ann. Appl. Biol. 1991, 118, 339–349. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate (Putative Species) | Sequence Length | Betanucleorhabdovirus Reads (% of Total #) | Reads Coverage |
|---|---|---|---|
| B160-295 (SaBNV5) | 13,527 | 22,782 (0.018%) | 227.4 |
| B160-296 (SaBNV5) | 13,524 | 64,643 (0.050%) | 647.9 |
| B160-297 (SaBNV3) | 13,508 | 88,653 (0.069%) | 892.1 |
| B160-298 cg (SaBNV1) | 13,488 | 570,049 (0.444%) | 5770.2 |
| B160-299 (SaBNV4) | 13,486 | 87,614 (0.068%) | 883.8 |
| B160-300 (SaBNV2) | 13,453 | 43,583 (0.034%) | 441.3 |
| B160-301 (SaBNV2) | 13,448 | 293,265 (0.229%) | 2962.1 |
| B160-302 cg (SaBNV2) | 13,458 | 240,413 (0.187%) | 2425.2 |
| B78-27 (SaBNV5) | 13,524 | 269,171 (0.233%) | 2824.0 |
| B78-28 (SaBNV3) | 13,518 | 268,699 (0.232%) | 3141.6 |
| B78-29 (SaBNV4) | 13,482 | 323,456 (0.280%) | 3426.7 |
| B78-30 (SaBNV2) | 13,438 | 13,584 (0.012%) | 143.4 |
| B42 cg (SaBNV3) | 13,521 | 4995 (0.003%) | 53.9 |
| N Gene | P Gene | P3 Gene | M Gene | G Gene | L Gene | Total Length | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate | nt Position | Length | nt Position | Length | nt Position | Length | nt Position | Length | nt Position | Length | nt Position | Length | |
| B160-298 | 182–1555 | 1374 | 1639–2670 | 1032 | 2775–3752 | 978 | 3934–4767 | 834 | 4943–6826 | 1884 | 7007–13,327 | 6321 | 13,488 |
| B78-30 | 168–1541 | 1374 | 1623–2654 | 1032 | 2765–3742 | 978 | 3909–4733 | 825 | 4906–6789 | 1884 | 6969–13,289 | 6321 | 13,438 |
| B160-302 | 179–1552 | 1634–2665 | 2776–3753 | 3920–4744 | 4917–6800 | 6980–13,300 | 13,458 | ||||||
| B160-300 | 174–1547 | 1629–2660 | 2770–3747 | 3914–4747 | 834 | 4911–6794 | 6975–13,295 | 13,453 | |||||
| B160-301 | 173–1546 | 1628–2659 | 2770–3747 | 3914–4747 | 4900–6783 | 6964–13,284 | 13,448 | ||||||
| B160-297 | 171–1544 | 1374 | 1627–2658 | 1032 | 2777–3754 | 978 | 3962–4798 | 837 | 4968–6851 | 1884 | 7030–13,350 | 6321 | 13,508 |
| B42 | 177–1550 | 1633–2664 | 2783–3760 | 3968–4804 | 4974–6857 | 7036–13,356 | 13,521 | ||||||
| B78-28 | 171–1544 | 1627–2658 | 2779–3756 | 3964–4800 | 4970–6853 | 7032–13,352 | 13,518 | ||||||
| B78-29 | 170–1543 | 1374 | 1626–2657 | 1032 | 2780–3757 | 978 | 3940–4776 | 837 | 4939–6828 | 1890 | 7006–13,326 | 6321 | 13,482 |
| B160-299 | 170–1543 | 1626–2657 | 2780–3757 | 3940–4776 | 4942–6831 | 7010–13,330 | 13,486 | ||||||
| B78-27 | 171–1544 | 1374 | 1631–2662 | 1032 | 2796–3773 | 978 | 3963–4811 | 849 | 4969–6858 | 1890 | 7040–13,360 | 6321 | 13,524 |
| B160-296 | 171–1544 | 1631–2662 | 2796–3773 | 3963–4811 | 4969–6858 | 7040–13,360 | 13,524 | ||||||
| B160-295 | 170–1543 | 1630–2661 | 2790–3767 | 3957–4805 | 4963–6852 | 7041–13,361 | 13,527 | ||||||
| B160-298 (PP711309) | TBRV1 (OL472119) | TBRV2 (OL441765) | DYVV (KM823531) | BmV2 (BK014480) | ZPNRV (MH323437) | SYVV (MT185675) | ||
|---|---|---|---|---|---|---|---|---|
| 1 | B160-298 | n/c | 64.7/70.4 | 65.1/70.2 | 53.3/48.4 | 52.9/48.1 | 52.7/48.0 | 50.5/45.5 |
| 2 | B78-30 | 69.5/79.4 | 64.4/69.7 | 65.6/70.2 | 52.9/48.0 | 53.3/48.3 | 52.7/48.3 | 50.6/45.3 |
| B160-302 | 69.4/79.5 | 64.4/69.8 | 65.5/70.2 | 53.1/48.1 | 53.3/48.3 | 52.8/48.2 | 50.7/45.4 | |
| B160-301 | 69.7/79.3 | 64.6/69.7 | 65.9/70.0 | 53.1/48.0 | 53.1/48.5 | 52.9/48.1 | 50.7/45.3 | |
| B160-300 | 69.8/79.0 | 64.6/70.2 | 65.3/70.3 | 53.4/47.7 | 53.0/48.2 | 52.8/48.0 | 50.4/44.9 | |
| 3 | B160-297 | 69.8/79.5 | 65.0/70.5 | 65.0/70.4 | 53.7/48.4 | 53.4/48.2 | 53.1/48.4 | 50.8/45.2 |
| B42 | 70.0/79.4 | 65.2/70.4 | 65.2/70.3 | 53.7/48.4 | 53.4/48.2 | 53.1/48.4 | 50.8/45.2 | |
| B78-28 | 69.9/79.5 | 65.1/70.6 | 64.8/70.4 | 53.8/48.4 | 53.4/48.1 | 53.0/48.5 | 50.6/45.2 | |
| 4 | B78-29 | 70.0/80.0 | 64.8/70.6 | 65.2/70.7 | 53.3/48.5 | 53.8/48.7 | 52.7/48.4 | 50.5/45.4 |
| B160-299 | 69.9/79.9 | 64.7/70.4 | 65.3/70.6 | 53.3/48.5 | 53.8/48.7 | 52.8/48.4 | 50.7/45.5 | |
| 5 | B78-27 | 70.3/81.0 | 65.2/70.2 | 65.2/70.2 | 53.2/48.3 | 53.3/48.2 | 52.6/48.4 | 50.3/45.3 |
| B160-296 | 70.4/80.9 | 65.2/70.3 | 65.3/70.2 | 53.3/48.3 | 53.4/48.1 | 52.8/48.4 | 50.2/45.3 | |
| B160-295 | 70.2/80.4 | 65.2/70.5 | 65.1/70.0 | 53.2/48.2 | 53.0/48.2 | 52.6/48.6 | 49.9/45.3 |
| dS/dN | 1 | 2 | 3 | 4 | 5 | All | Ps | π | D |
|---|---|---|---|---|---|---|---|---|---|
| N | n/c | 0.431/0.008 | 0.142/0.002 | 0.108/0.002 | 0.366/0.007 | 0.726/0.029 | 0.107 | 0.040 | 0.754 |
| P | n/c | 0.393/0.052 | 0.156/0.0181 | 0.741/0.153 | 0.546/0.095 | 0.694/0.148 | 0.351 | 0.188 | 3.039 |
| P3 | n/c | 0.424/0.006 | 0.138/0.001 | 0.148/0.001 | 0.323/0.008 | 0.717/0.037 | 0.122 | 0.047 | 0.888 |
| M | n/c | 0.375/0.024 | 0.148/0.004 | 0.115/0.013 | 0.309/0.017 | 0.672/0.108 | 0.319 | 0.147 | 1.955 |
| G | n/c | 0.429/0.024 | 0.142/0.006 | 0.113/0.004 | 0.378/0.024 | 0.718/0.096 | 0.311 | 0.148 | 2.181 |
| L | n/c | 0.413/0.026 | 0.144/0.009 | 0.122/0.006 | 0.365/0.025 | 0.510/0.110 | 0.472 | 0.233 | 2.446 |
| CDS | n/c | 0.393/0.052 | 0.156/0.018 | 0.100/0.006 | 0.403/0.045 | 0.694/0.148 | 0.292 | 0.136 | 2.076 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šafářová, D.; Candresse, T.; Veselská, J.; Navrátil, M. Novel Betanucleorhabdoviruses Infecting Elderberry (Sambucus nigra L.): Genome Characterization and Genetic Variability. Pathogens 2024, 13, 445. https://doi.org/10.3390/pathogens13060445
Šafářová D, Candresse T, Veselská J, Navrátil M. Novel Betanucleorhabdoviruses Infecting Elderberry (Sambucus nigra L.): Genome Characterization and Genetic Variability. Pathogens. 2024; 13(6):445. https://doi.org/10.3390/pathogens13060445
Chicago/Turabian StyleŠafářová, Dana, Thierry Candresse, Jana Veselská, and Milan Navrátil. 2024. "Novel Betanucleorhabdoviruses Infecting Elderberry (Sambucus nigra L.): Genome Characterization and Genetic Variability" Pathogens 13, no. 6: 445. https://doi.org/10.3390/pathogens13060445
APA StyleŠafářová, D., Candresse, T., Veselská, J., & Navrátil, M. (2024). Novel Betanucleorhabdoviruses Infecting Elderberry (Sambucus nigra L.): Genome Characterization and Genetic Variability. Pathogens, 13(6), 445. https://doi.org/10.3390/pathogens13060445