
Advanced Protocol for Molecular Characterization of Viral Genome in Fission Yeast (Schizosaccharomyces pombe)



Abstract
:1. Introduction
2. Materials
2.1. Fission Yeast Strains, Plasmids, and Molecular Cloning
- PCR primers specific to the viral gene for amplification;
- PCR kit containing DNA polymerase and nucleotides (dNTPs);
- DNA gel extraction kit for purification of PCR products;
- Restriction enzymes and respective buffers specific to the cloning strategy (e.g., for linearizing the plasmid vector and digesting the insert, see Figure 2);
- DNA ligase for joining viral gene inserts with plasmid DNA;
- Gel electrophoresis apparatus for analyzing DNA fragments;
- DNA ladder or marker for size determination;
- Agarose gel and TAE or TBE buffer for gel electrophoresis;
- UV transilluminator or an imaging system for visualizing DNA bands;
- A wild-type fission yeast strain such as SP223 or 972 for plasmid DNA transformation;
- Selective media for fission yeast (e.g., minimal medium supplemented with appropriate nutrients; also refer to Section 2.2. Fission Yeast Growth Media).
2.2. Fission Yeast Growth Media
- Standard Yeast Extract with Supplements (YES) medium: 0.5% (w/v) yeast extract, 3.0% (w/v) glucose, supplements: 225 mg/L adenine (Ade), leucine (Leu) and uracil (Ura) (see Note 1).
- Edinburgh Minimal Medium (EMM): 14.7 mM potassium hydrogen phthalate, 15.5 mM Na2HPO4, 93.5 mM NH4Cl, 2.0% (w/v) glucose, 20 mL/L 50× salt stock, 1 mL/L 1000× vitamin stock, 0.1 mL/L 10,000× mineral stock (see Note 2).
- 50× Salt Stock: 0.26 M MgCl2, 5 mM CaCl2, 0.67 M KCl, 14.1 mM Na2SO4.
- 1000× Vitamin Stock: 4.20 mM pantothenic acid, 81.2 mM nicotinic acid, 55.5 mM inositol, 40.8 μM biotin.
- 10,000× Mineral Stock: 80.9 mM boric acid, 23.7 mM MnSO4, 13.9 mM ZnSO4∙7H2O, 7.40 mM FeCl2∙6H2O, 2.47 mM molybdic acid, 6.02 mM KI, 1.60 mM CuSO4∙5H2O, 47.6 mM citric acid.
- Pombe Minimal Glutamate (PMG) Medium: potassium hydrogen phthalate 3 g/L (14.7 mM); Na2HPO4 2.2 g/L (15.5 mM); L-glutamic acid, monosodium salt (3.75 g/L); glucose 20 g/L (2% w/v); salt mix 20 mL/L; vitamin mix 1 mL/L; mineral mix 0.1 mL/L (see Note 3).
- For solid fission yeast media, add 2% (w/v) agarose.
- Luria–Bertani (LB) Medium: 1% (w/v) tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl (see Note 4).
- For solid LB media, add 1.5% (w/v) agarose.
2.3. Inducible Gene Expression in Fission Yeast
2.4. Determination of Subcellular Localization
- Microscope slides and coverslips.
- DAPI staining stock solution in water (1 mg/mL). Store at −20 °C. Use 1 µg/mL in the final concentration.
- Hoechst 33342 staining stock solution in water (1 mg/mL). Store at −20 °C. Use 1 µg/mL in the final concentration.
- Sodium citrate (pH 7.0) stock solution (500 mM). Store at room temperature. Use 50 mM in the final concentration.
- Propidium iodine (PI) stock solution in water (4 mg/mL). Store in the dark at −20 °C. Use 0.1 mg/mL in the final concentration.
- RNase A (10 mg/mL; boil 10 min, cool to RT, filter and store at −20 °C). Use 0.1 mg/mL in the final concentration.
- Trypan Blue solution (0.4% (w/v)). Store at room temperature. Use 0.2% (w/v) in the final concentration.
- NH4Cl (1.4 M). Store at room temperature.
- Thiamine stock solution in distilled water (20 mM). Store at −20 °C.
- GH solution (2% (w/v) D-(+)-glucose +10 mM Na-HEPES, pH 7.2). Store at room temperature.
- FUN-1 working solution (80 μM). Store at −20 °C [27].
- Leica DM fluorescent microscope with 11001v2 long-path Chroma filter cube.
2.5. Western Blot Analysis of Fission Yeast Proteins
- Protein samples from fission yeast (cell lysates or purified proteins).
- SDS-PAGE gel or other appropriate gel system for protein separation.
- Transfer buffer suitable for fission yeast proteins (e.g., tris-glycine buffer).
- Nitrocellulose or PVDF membrane.
- Transfer apparatus (e.g., wet or semi-dry transfer system).
- Blocking buffer compatible with fission yeast proteins (e.g., 5% (w/v) non-fat dry milk or BSA in PBS-T).
- Primary antibody specific to the fission yeast protein of interest.
- Secondary antibody conjugated with HRP (horseradish peroxidase) compatible with fission yeast proteins.
- Chemiluminescent substrate suitable for detection of fission yeast proteins (e.g., ECL).
- Film or chemiluminescent imaging system.
- Washing buffer compatible with fission yeast proteins (e.g., PBS-T).
- Electrophoresis apparatus suitable for running SDS-PAGE gels.
- Protein marker or ladder.
- Blocking agent (e.g., non-fat dry milk or BSA).
- Gel documentation system or imaging equipment for visualization and analysis.
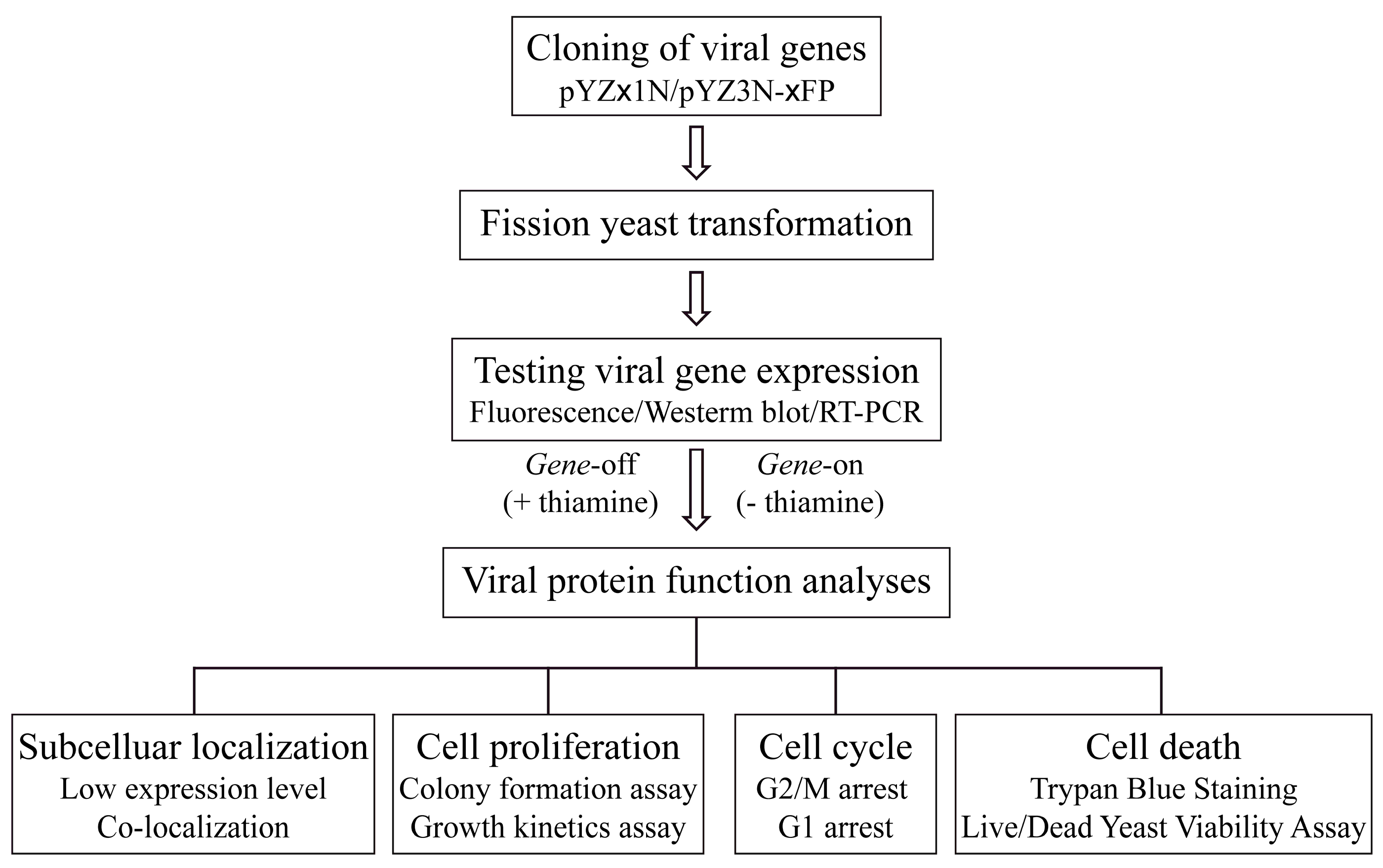
3. Methods
3.1. Genome-Wide Molecular Cloning of Viral Proteins
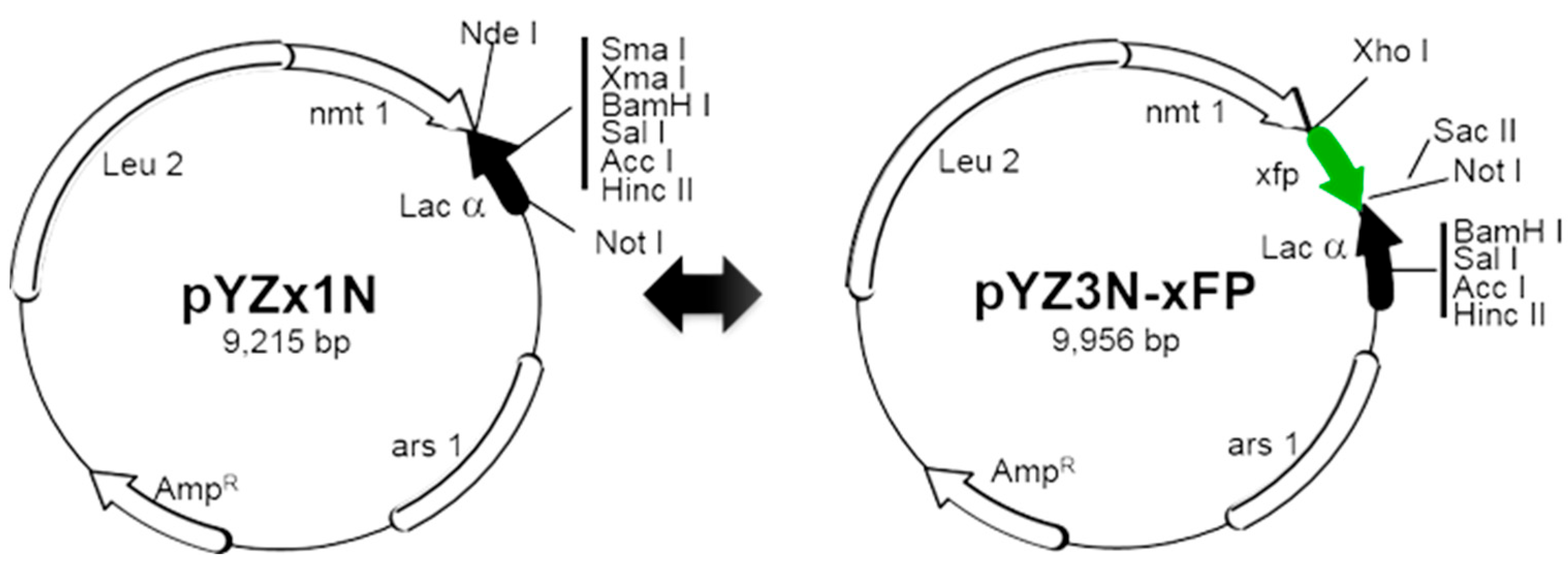
- Viral genes are inserted into the destination vector using the traditional cloning method, which involves the use of restriction enzymes to cut DNA and ligase to join DNA. For instance, to clone a viral gene into the pYZ1N plasmid, first amplify the gene via PCR with a forward primer containing an NdeI restriction site and a reverse primer containing the stop codon, along with one of the following single-cutter restriction sites: NdeI, XmaI, XmaI, BamHI, SalI, or NotI on the pYZ1N plasmid (Figure S1). After amplification, ligate the digested PCR products into the vector following the same digestion. Note that the NdeI site already includes a start codon (ATG) and using the NotI site will remove the entire Lac fragment preceding the Nmt1 terminator sequence (see Note 11).
- To clone a C-terminus GFP-tagged viral gene into the pYZ3N-GFP, utilize the single-cutter XhoI site (Figure S2). The forward primer should include the start codon, while the reverse primer should remove the stop codon. Note the presence of an in-frame stop codon at the end of GFP immediately following the SacII/NotI sites.
- To clone an N-terminus GFP-tagged viral gene into the pY3N-GFP, employ the single-cutter SacII or NotI restriction enzyme (Figure S2). Employing SacII/NotI and XmaI/XmaI restriction enzymes for viral gene insertion will result in the removal of the full Lac fragment from the pYZ3N-GFP plasmid (Figure S2). This ensures the proper orientation of the N-terminus tagged viral gene. In this scenario, the stop side primer must also contain an in-frame stop codon.
- To clone a viral gene fused with GFP directly into the pYZx1N plasmid, overlapping PCR or three-fragment ligation could be employed. Amplify the viral gene and xFP separately via PCR, using viral genome for the viral gene and any DNA source for xFP amplification. Similarly to Section 1, utilize the combination of NdeI with NdeI, XmaI, XmaI, BamHI, SalI, or NotI (Figure S1).
- Recent advances in molecular cloning methodologies utilize other properties of DNA polymerase, such as exonuclease activity, along with DNA homologous recombination to simplify the cloning process could also be used. Techniques like NEBuilder HiFi DNA Assembly, Gibson Assembly [31], and Takara in-fusion cloning can be employed to create these constructs. Taking NEBuilder HiFi DNA Assembly as an example, to clone a C-terminus xFP-tagged viral gene into the pYZ1N plasmid, amplify the viral gene using a forward primer (VG-F) containing a 5′-end overlapping sequence of 20–30 bp with the nmt promoter of the pYZ1N plasmid and a 3′-end overlapping sequence of 20 bp with the viral gene start side (containing the ATG start codon), and a reverse primer (VG-R) containing a 5′-end overlapping sequence of 15–30 bp with the xFP start side and a 3′-end overlapping sequence of 20 bp with the viral gene end side (excluding the stop codon). Amplify xFP using a forward primer (xFP-F) containing a 5′-end overlapping sequence of 15–30 bp with the viral gene end part (excluding the stop codon) and a 3′-end overlapping sequence of 20 bp with the xFP gene start side, and a reverse primer (xFP-R) containing a 5′-end overlapping sequence of 20–30 bp with the nmt terminator of the pYZ1N plasmid and a 3′-end overlapping sequence of 20 bp with the xFP end side (including the stop codon). After digesting pYZ1N with NdeI-NotI, the three fragments (digested plasmid plus two PCR products) can be assembled easily using the “NEBuilder® HiFi DNA Assembly” kit in one step. See the flanking sequences of pYZ1N in Figure S1.
- Verification of the insert and its orientation is crucial. Firstly, this can be achieved by PCR or restriction digestion using one of the described cloning methods and an inner restriction site if a single restriction site was used for insertion. Subsequently, the final plasmid should be further confirmed by Sanger sequencing or whole plasmid sequencing.
- Carefully design primers for gene amplification to ensure that the viral gene and/or the xFP ORF are in frame.
3.2. Recombinant DNA Transformation and Inducible Viral Gene Expression
- Transformants are selected based on either Leu or Ura auxotrophy on selective EMM, depending on whether the plasmid carries a LEU2 or URA4 gene.
- Successful transformation of the respective viral-gene-containing plasmid is confirmed through single-colony PCR [15].
- Conditional induction of viral gene expression is achieved by thiamine removal from the growth medium, facilitating the measurement of viral-gene-specific activities over time [13,22]. Importantly, all viral activities can be concurrently measured under identical inducible conditions, expediting the functional characterization of the viral genome of interest.
- Verification of viral gene expression can be accomplished in the following ways:
- Observing xFP production using a fluorescent microscope;
- Measuring viral protein production by Western blot analysis with a monoclonal antibody;
- Measuring mRNA levels using quantitative RT-PCR.
- To assess viral-gene-specific activities, a single yeast colony containing a specific viral-gene-containing plasmid is cultured to the logarithmic (log) phase in special liquid EMM supplemented with 20 µM of thiamine.
- Cells are subsequently harvested and washed three times with distilled water to eliminate thiamine.
- Finally, 2 × 105 cells/mL, or a cell concentration that is determined empirically for optimal gene expression, are re-inoculated into fresh, specific liquid EMM without thiamine to induce gene expression (Gene-on) or with thiamine to suppress gene expression (Gene-off), which serves as the control (see Notes 5 and 6).
3.3. Determination of Subcellular Localization of Viral Proteins
- The viral proteins fused to GFP are expressed as described above, and their subcellular locations are determined using fluorescent microscopy. To mitigate artifacts stemming from high-level expression of the viral proteins, 10 nM of thiamine is added to the EMM to reduce the level of viral protein expression [12,32] (refer to Note 6, 7). Typically, the subcellular localization of each viral protein is visualized within 20 h after gene induction using fluorescent microscopy.
- To aid in determining the subcellular location of a protein, a fluorescent DNA dye, 4′,6′ diamino-2-phenylindole (DAPI) for fixed cells or Hoechst 33342 for live or fixed cells, is commonly used to stain nuclei, enabling differentiation of whether the viral protein is associated with the nucleus or other subcellular compartments.
- For Hoechst 33342 or DAPI staining, 2–5 µL of the viral-gene-expressing cell suspension is pipetted onto a glass slide. The cells are heat-fixed for 1 min at 70 °C on a hot plate. The slide is then cooled for a few seconds before adding Hoechst 33342 or DAPI (1 µg/mL). A coverslip is placed over the cells. Cells expressing GFP-fused viral proteins and stained with Hoechst 33342 or DAPI are observed under fluorescence microscopy (refer to Figure 3).
- To validate the specific subcellular location of a viral protein, fission yeast cellular proteins known to localize specifically in, for example, nuclear membrane localization of HIV-1 Vpr protein (Figure 3A) [33], lysosomal localization of SARS-CoV-2 ORF3a protein (Figure 3B) [16,34], and various ZIKV proteins localized in the endoplasmic reticulum (Gpi16), Golgi (Ynd1), and mitochondria (Rsm10) [14] can be utilized for comparison (Figure 3Ca) (refer to Note 8).
- Image analysis is conducted using a Leica DMR fluorescence microscope equipped with a high-performance charge-coupled device camera (Hamamatsu) and Open-Lab software (Improvision, Inc., Lexington, MA, USA). For GFP observation, a Leica L5 filter with excitatiominn at 480/40 nm and emission at 527/30 nm is used. A Leica YFP filter with excitation at 500/20 nm and emission at 535/30 nm is employed for YFP observation. Note that the spectrum of GFP and YFP overlaps. Therefore, we cannot distinguish these two proteins under the GFP/YFP filter. However, we could use the deduction method to detect the GFP signal under the CFP filter (Figure 3Bb). For CFP observation, a specific Leica CFP filter with excitation at 436/20 nm and emission at 480/40 nm is utilized (refer to Note 9).

3.4. Measurement of Cell Proliferation
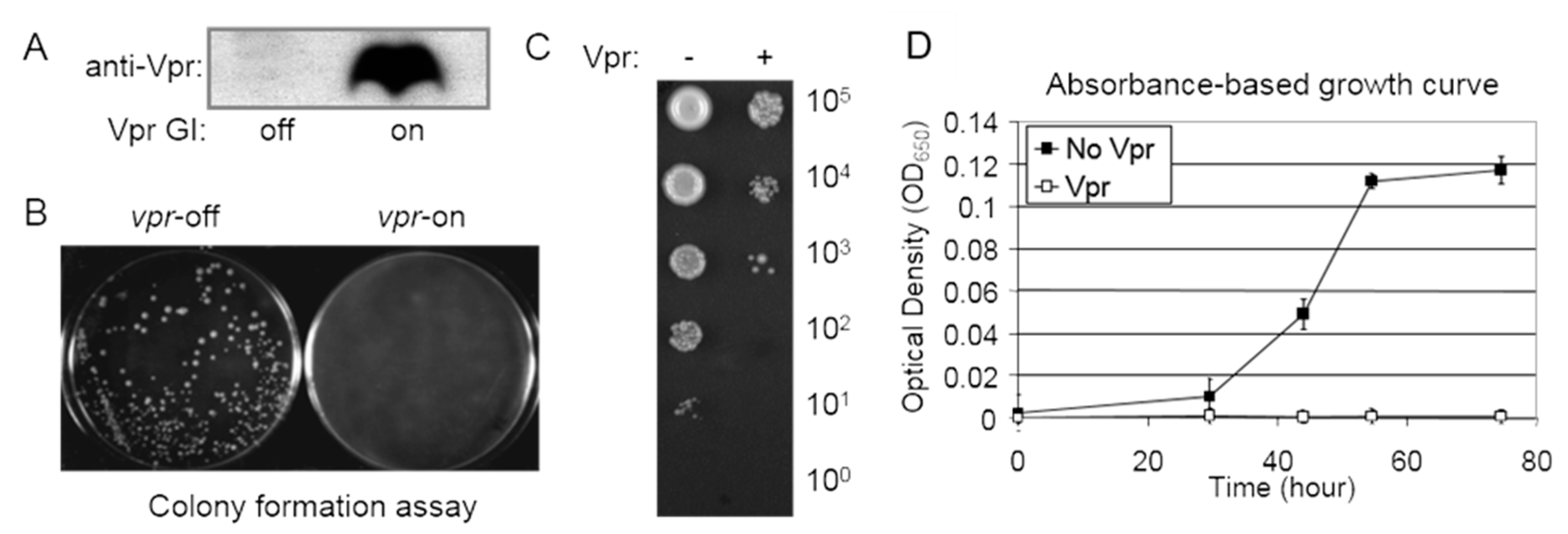
- A single yeast colony containing a viral-gene-containing plasmid is selected from the selective EMM plate and cultured overnight in specific liquid EMM supplemented with thiamine. The following day, 1 mL of mid-log-phase culture is centrifuged, washed three times with distilled water, and the cells are resuspended in an appropriate volume of EMM. Approximately 100 µL of liquid cultures containing around 1000 cells is spread onto selective EMM agar plates with and without thiamine. Inducible protein production can be accessed via Western blot analysis (Figure 4A). The agar plates are then incubated at 30 °C for 4–6 days to observe the presence or absence, as well as the sizes, of forming colonies. The absence of colonies on the agar plates may indicate a potential cell-killing effect, while smaller colony sizes compared to the control typically suggest potential growth restriction (Figure 4B).
- A semi-quantitative colony assay is employed to further assess the extent of growth inhibition presumably observed in the colony formation assay described above.
- Using the same preparation procedure as described above, approximately 1–5 µL (instead of 100 µL) of liquid EMM cultures with serial 10-fold dilutions (ranging from 106 to 100 cells) is spotted onto selective EMM agar plates. The plates are then incubated at 30 °C for 3–6 days to observe the level of colony formation dilution and the number of cells within each colony (at low dilutions) as a semi-quantitative indicator of the viral effect on cellular growth or cytotoxicity (see an example in Figure 4C).
- For quantitative measurements of growth inhibition, a growth kinetics assay is employed (Figure 4D). Specifically, liquid cell cultures are grown in a 96-well microtiter plate containing 100 µL of selective EMM. Cell cultures are prepared as described above and incubated at 30 °C in a moist incubator. Cellular growth is monitored at OD650 over the specified time period using a spectrophotometer (Figure 4D) after shaking the cultures in the 96-well plate.
3.5. Cell Cycle Profiling
- Cells containing the pYZ1N viral gene are cultured to the stationary phase in 5 mL of EMM containing thiamine, with constant shaking at 30 °C. A 1 mL aliquot of the culture is collected, washed three times with distilled water to eliminate thiamine, and re-inoculated into 5 mL of culture medium at a concentration of 2 × 105 cells per mL with or without thiamine.
- Cells are harvested in approximately 48 h. Spin down 107 cells from the liquid culture at 2000 revolutions per minute (rpm) for 5 min, fix the cells with 1 mL of 70% cold ethanol, and store at 4 °C.
- Before flow cytometry analysis, take 200 µL of cells and add them to 2 mL of 50 mM sodium citrate (pH 7.0) in a 5 mL Falcon tube, mix well, and centrifuge at 2000 rpm for 5 min. Treat the cells with RNase A (0.1 mg/mL) in 50 mM sodium citrate for 2 h at 37 °C, and then stain with propidium iodide (PI, 4 µg/mL) on ice for at least 1 h.
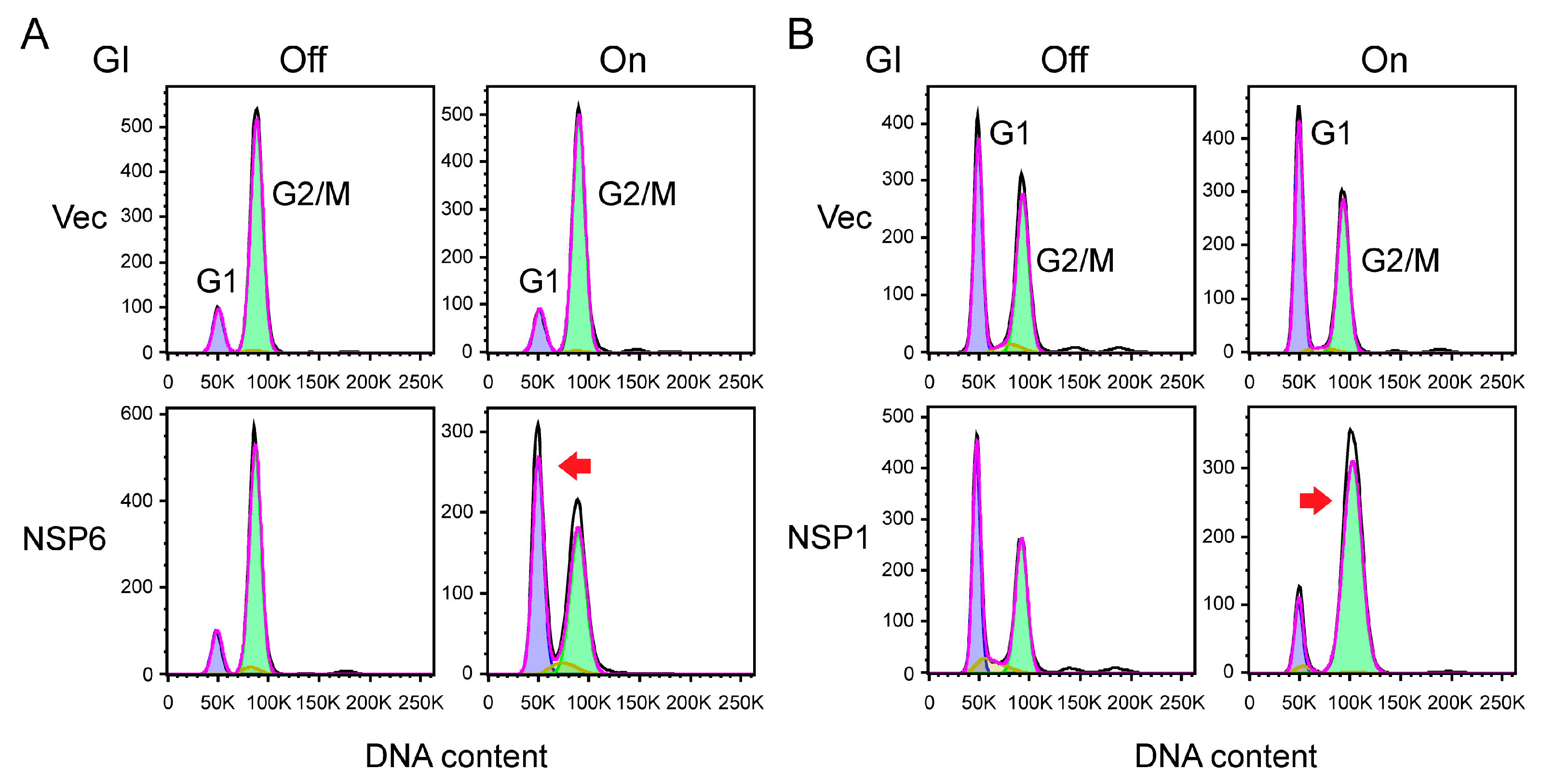
- To test whether production of a viral protein in fission yeast induces cell cycle G1 arrest, fission yeast cells that are cultured in regular EMM can be used in flow cytometric analysis, as fission yeast cells primarily reside in the G2/M phase of the cell cycle [33,41] (refer to Figure 5A, top, our unpublished data). Here, we show that expression of SARS-CoV-2 NSP6 protein significantly shifts the cell cycle profile from the G2 phase to the G1 phase (Figure 5A, top, our unpublished data).
- To evaluate whether viral protein production causes cell cycle G2/M arrest, fission yeast cells need to first be cultured in low-nitrogen (LN) medium containing 2.5 mM NH4Cl, which enriches fission yeast cells in the G1 phase of the cell cycle [12,33] (refer to Figure 5B, top, our unpublished data). As we show here, the expression of SARS-CoV-2 NSP1 protein significantly shifts the cell cycle profile from the G1 phase to the G2/M phase (Figure 5B, bottom, our unpublished data).
- Both EMM and LN media can be utilized to measure the effect on the S phase.
- The DNA content is analyzed on a FACSCanto II (Becton Dickinson) using the FACS DIVA 6.3 software (Becton Dickinson) or whatever software is available. Ten thousand events are collected, and the level of DNA content corresponding to cells in G1, G2/M, or S is determined as the FL2 parameter (FL-2 measures the amount of PI fluorescence emitted through a 585 nm band-pass filter). When PI binds to nucleic acid, it has an excitation maximum of ~535 nm and an emission maximum of ~615 nm.
3.6. Measurement of Cell Death
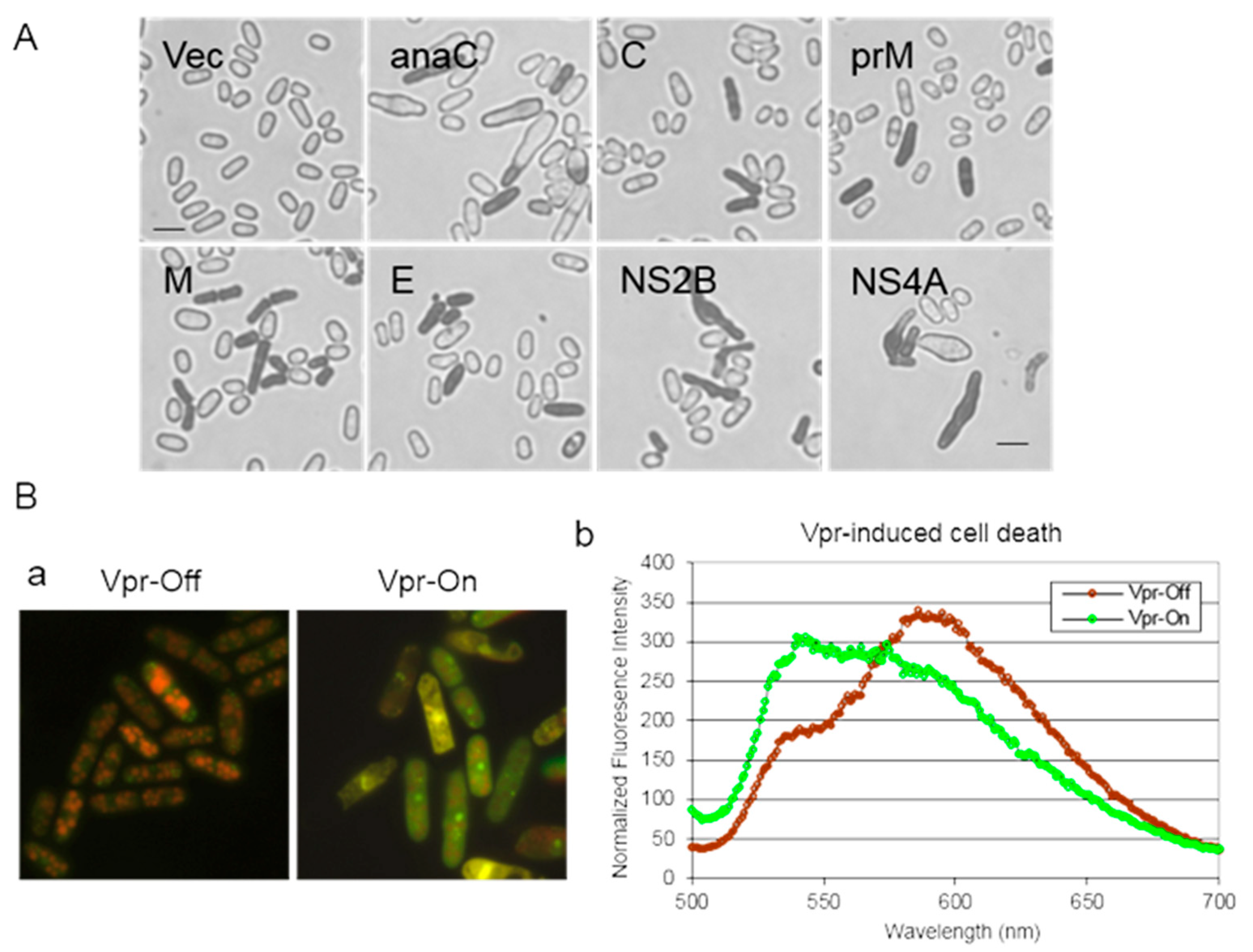
- To quantify the number of dead cells, cell cultures are prepared as described above. The percentage of cell death induced by a viral protein is monitored over time following viral gene induction. Trypan Blue (Thermo Fisher Scientific, Waltham, MA, USA) at a final concentration of 0.2% (w/v) is introduced into the cell culture. Trypan Blue, a diazo dye, exclusively stains dead cells; live cells with intact membranes remain unstained. Consequently, the percentage of cell death is determined by counting the number of Trypan Blue-stained cells relative to the total number of cells counted (refer to Figure 6A).
- Cell viability can also be assessed using a commercial Live/Dead Yeast Viability Assay (Cat. No. L-7009; Invitrogen, Carlsbad, CA, USA), adapted for fission yeast [27,39,43]. This assay measures cell viability by monitoring intracellular metabolic activity through FUN-1 staining. Metabolically active cells convert the yellow-green, fluorescent intracellular FUN-1 into red-orange intra-vacuolar structures, emitting fluorescence at 590 nm. Metabolically inert or dead cells exhibit bright, diffuse, green–yellow fluorescence at ~540 nm (Figure 6B).
- Briefly, thiamine is removed from a log-phase cell culture as described previously. Cells are then diluted to a concentration of 5 × 104 cells/mL and resuspended in EMM supplemented with or without thiamine to suppress or induce viral gene expression, respectively. The cell culture is incubated at 30 °C with constant shaking at 300 rpm and collected over time.
- The cell culture is resuspended in GH solution (2% (w/v) D-(+)-glucose +10 mM Na-HEPES, pH 7.2). A 50 µL aliquot of FUN-1 solution (80 µM) is added to an equal volume of cell suspension. The suspension is further incubated at 30 °C for 45 min. Approximately 3 µL of the suspension is applied onto a glass slide and covered with a coverslip. Cell viability is examined using a Leica DM fluorescent microscope equipped with a 11001v2 long-path Chroma filter cube. Typically, actively respiring cells are clearly marked with orange–red fluorescent structures at a maximum wavelength of approximately 590 nm, while metabolically inert or dead cells exhibit bright, diffuse, green–yellow fluorescence at a maximum wavelength of approximately 540 nm [27,39,43]. FUN1-stained cell images are captured using red (N2.1, emission LP 590 nm) and green (YFP, emission 535/30 nm) filters. Final images are generated by merging fluorescence (refer to Figure 6B).
4. Notes
- The YES medium is the most commonly used complete and rich medium and is normally used to grow fission yeast cells without selection.
- EMM is typically used to select for the presence of a plasmid that carries either a LEU2 gene or URA4 gene in fission yeast cells that are deficient in leu1-32 or ura4-294 such as SP223. Supplement with adenine, uracil, or leucine (225 mg/L) to complement the corresponding auxotrophic mutants of the yeast strain. Normally, 20 μM of thiamine is used to suppress the nmt1 promoter.
- As only a limited number of auxotrophic nutrient deficiency markers are available for selection, antibiotic resistance markers are good alternatives. However, antibiotic resistance markers typically work well in complete rich medium but not in defined EMM. We found that Zeocin works well in PMB medium to select and maintain bleMX6 resistance in conjunction with a LEU2 auxotrophic marker in fission yeast [21].
- Supplement LB medium with ampicillin (100 μg/mL) to select for the presence of the plasmid. This medium is used for growing bacterium E. coli Top 10 or DH5α cells and for plasmid DNA transformation.
- The unique features of this vector system are as follows: It simplifies the gene cloning process into two fission yeast pYZ vectors in a sequential manner. All gene cloning occurs unidirectionally, ensuring positive identification of gene insertions. An inducible gene transcriptional nmt1 promoter [11,13,22] is utilized to facilitate the determination of gene-specific effects. Three different strengths of the nmt1 promoter (high, intermediate, and low) are coupled with two distinct cell growth markers (leu2 and ura4) and one antibiotic selection marker (bleMX6) [11,13,21,22], enabling the testing of the gene expression at various levels. Three different fluorescent proteins (GFP, YFP, and CYP) are available for evaluating subcellular localization and co-localization of viral proteins within cells.
- Protein overproduction in fission yeast cells can lead to the formation of protein aggregates, potentially obscuring the true subcellular location of the protein of interest. To mitigate this artifact, 10 nM of thiamine is added to the EMM to reduce viral protein expression levels [15,44]. This precaution is particularly crucial when determining the subcellular location of a cellular protein that is naturally present in low quantities.
- Conversely, viral infection can result in high viremia, leading to the abundant production of viral proteins within cells. To simulate this scenario, each viral protein can be expressed over time using the full strength of the nmt1 promoter without thiamine. The impact of high viral protein expression levels on subcellular localization can then be documented and compared with low levels of expression [14].
- For measuring potential co-localization of two proteins, it is advisable to use two different fluorescent protein tags with non-overlapping excitation and/or emission spectra. YFP and CFP are commonly employed for this purpose. While GFP is widely used, its excitation spectrum overlaps with both YFP and CFP. Therefore, GFP is not ideal for co-localization studies. However, since YFP cannot be excited under the excitation spectrum of the CFP filter, which has an excitation of 436/20 nm and emission of 480/40 nm, no YFP signal is detected under the CFP filter. Thus, it can be used to differentiate the YFP signal from GFP [14].
- Other fluorescent microscopes equipped with suitable filter cubes can also be utilized to observe these fluorescent proteins.
- It should be emphasized that utilizing fission yeast as a surrogate for virologic studies certainly has its limitations, as it may not fully represent the complex cellular interactions involving in a viral protein of interest. Therefore, subsequent validation of identified viral protein properties in the host cell and within the context of viral infection is imperative. Also, note that this protocol is tailored for investigating viral proteins in fission yeast. All molecular constructs described here are intended for use in fission yeast studies only. Consequently, the viral gene-of-interest identified within the fission yeast system must be cloned into a mammalian expression vector for translational studies in mammalian cells.
- NEBuilder® HiFi DNA Assembly offers a streamlined process for inserting one or multiple DNA fragments into the pYZ1N plasmid in a single step, ensuring proper orientation (Figure S1). For a single insertion, only the linearized pYZ1N and the PCR product of the viral gene are required. The forward primer of the viral gene (primer VG-F) should contain a 5′-end sequence overlapping by 20–30 bp with the nmt promoter of the pYZ1N plasmid and a 3′-end sequence overlapping by 20 bp with the viral gene start site (containing the ATG start codon). Similarly, the reverse primer of the viral gene (primer VG-R) should include a 5′-end sequence overlapping by 20–30 bp with the nmt terminator of the pYZ1N plasmid and a 3′-end sequence overlapping by 20 bp with the viral gene end site (including the stop codon).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartwell, L.H. Yeast and cancer. Biosci. Rep. 2004, 24, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. A prize for proliferation. Cell 2001, 107, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem 2002, 3, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. From fission to fusion: A perspective on the research that won the Nobel Prize in Physiology or Medicine, 2013. J. Biosci. 2014, 39, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y. Yeast for virus research. Microb. Cell 2017, 4, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hom, K.; Zhang, C.; Nasr, M.; Gerzanich, V.; Zhang, Y.; Tang, Q.; Xue, F.; Simard, J.M.; Zhao, R.Y. SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects. Pathogens 2024, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Elder, R.T. Yeast perspectives on HIV-1 Vpr. Front. Biosci. 2000, 5, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Elder, R.T. Viral infections and cell cycle G2/M regulation. Cell Res. 2005, 15, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Andreola, M.L.; Litvak, S. Yeast and the AIDS virus: The odd couple. J. Biomed. Biotechnol. 2012, 2012, 549020. [Google Scholar] [CrossRef]
- Lista, M.J.; Voisset, C.; Contesse, M.A.; Friocourt, G.; Daskalogianni, C.; Bihel, F.; Fahraeus, R.; Blondel, M. The long-lasting love affair between the budding yeast Saccharomyces cerevisiae and the Epstein-Barr virus. Biotechnol. J. 2015, 10, 1670–1681. [Google Scholar] [CrossRef]
- Zhao, Y.; Elder, R.T.; Chen, M.; Cao, J. Fission yeast expression vectors adapted for large scale cloning and GFP fusion with positive screening. BioTechniques 1998, 25, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, R.Y. Molecular Cloning and Characterization of Small Viral Genome in Fission Yeast. In Methods in Molecular Biology, 2018/02/10 ed.; Singleton, T., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1721, pp. 47–61. [Google Scholar]
- Maundrell, K. nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem. 1990, 265, 10857–10864. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385. [Google Scholar] [CrossRef] [PubMed]
- Nkeze, J.; Li, L.; Benko, Z.; Li, G.; Zhao, R.Y. Molecular characterization of HIV-1 genome in fission yeast Schizosaccharomyces pombe. Cell Biosci. 2015, 5, 47. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Q.; Cruz Cosme, R.S.; Gerzanich, V.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Genome-Wide Characterization of SARS-CoV-2 Cytopathogenic Proteins in the Search of Antiviral Targets. mBio 2022, 13, e0016922. [Google Scholar] [CrossRef]
- Jin, H.; Du, Z.; Zhang, Y.; Antal, J.; Xia, Z.; Wang, Y.; Gao, Y.; Zhao, X.; Han, X.; Cheng, Y.; et al. A distinct class of plant and animal viral proteins that disrupt mitosis by directly interrupting the mitotic entry switch Wee1-Cdc25-Cdk1. Sci. Adv. 2020, 6, eaba3418. [Google Scholar] [CrossRef] [PubMed]
- Rallis, C.; Bahler, J. Cell-based screens and phenomics with fission yeast. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Sahaya Glingston, R.; Yadav, J.; Rajpoot, J.; Joshi, N.; Nagotu, S. Contribution of yeast models to virus research. Appl. Microbiol. Biotechnol. 2021, 105, 4855–4878. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Klar, A.; Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991, 194, 795–823. [Google Scholar] [PubMed]
- Benko, Z.; Zhao, R.Y. Zeocin for selection of bleMX6 resistance in fission yeast. Biotechniques 2011, 51, 57–60. [Google Scholar] [CrossRef]
- Basi, G.; Schmid, E.; Maundrell, K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 1993, 123, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Forsburg, S.L.; Sherman, D.A. General purpose tagging vectors for fission yeast. Gene 1997, 191, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Siam, R.; Dolan, W.P.; Forsburg, S.L. Choosing and using Schizosaccharomyces pombe plasmids. Methods 2004, 33, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Forsburg, S.L. Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 1993, 21, 2955–2956. [Google Scholar] [CrossRef]
- Javerzat, J.P.; Cranston, G.; Allshire, R.C. Fission yeast genes which disrupt mitotic chromosome segregation when overexpressed. Nucleic Acids Res. 1996, 24, 4676–4683. [Google Scholar] [CrossRef]
- Benko, Z.; Elder, R.T.; Liang, D.; Zhao, R.R. Fission yeast as a HTS platform for molecular probes of HIV-1 Vpr-induced cell death. Int. J. High Throughput Screen. 2010, 1, 151–162. [Google Scholar]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Zhao, Y.; Lieberman, H.B. Schizosaccharomyces pombe: A model for molecular studies of eukaryotic genes. DNA Cell Biol. 1995, 14, 359–371. [Google Scholar] [CrossRef]
- Olsson, I.; Bjerling, P. Advancing our understanding of functional genome organisation through studies in the fission yeast. Curr. Genet. 2011, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Benko, Z.; Zhang, J.; Zhao, R.Y. Development of A Fission Yeast Cell-Based Platform for High Throughput Screening of HIV-1 Protease Inhibitors. Curr. HIV Res. 2019, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; O’Gorman, M.R.; Yu, M.; Yogev, R. Effect of human immunodeficiency virus type 1 protein R (vpr) gene expression on basic cellular function of fission yeast Schizosaccharomyces pombe. J. Virol. 1996, 70, 5821–5826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cruz-Cosme, R.; Zhang, C.; Liu, D.; Tang, Q.; Zhao, R.Y. Endoplasmic reticulum-associated SARS-CoV-2 ORF3a elicits heightened cytopathic effects despite robust ER-associated degradation. mBio 2024, 15, e0303023. [Google Scholar] [CrossRef]
- Xu, D.D.; Du, L.L. Fission Yeast Autophagy Machinery. Cells 2022, 11, 1086. [Google Scholar] [CrossRef]
- Cruz-Cosme, R.; Zhang, J.; Liu, D.; Mahase, V.; Sallapalli, B.T.; Chang, P.; Zhang, Y.; Teng, S.; Zhao, R.Y.; Tang, Q. A novel diG motif in ORF3a protein of SARS-CoV-2 for intracellular transport. Front. Cell Dev. Biol. 2022, 10, 1011221. [Google Scholar] [CrossRef] [PubMed]
- Vida, T.A.; Emr, S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995, 128, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Elder, R.T.; Yu, M.; O’Gorman, M.G.; Selig, L.; Benarous, R.; Yamamoto, A.; Zhao, Y. Mutational analysis of Vpr-induced G2 arrest, nuclear localization, and cell death in fission yeast. J. Virol. 1999, 73, 3236–3245. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, M.; Chen, M.; Elder, R.T.; Yamamoto, A.; Cao, J. Pleiotropic Effects of HIV-1 Protein R (Vpr) on Morphogenesis and Cell Survival in Fission Yeast and Antagonism by Pentoxifylline. Virology 1998, 246, 266–276. [Google Scholar] [CrossRef]
- Forsburg, S.L.; Nurse, P. Cell cycle regulation in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe. Annu. Rev. Cell Biol. 1991, 7, 227–256. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Kucsera, J.; Yarita, K.; Takeo, K. Simple detection method for distinguishing dead and living yeast colonies. J. Microbiol. Methods 2000, 41, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Benko, Z.; Elder, R.T.; Li, G.; Liang, D.; Zhao, R.Y. HIV-1 Protease in the Fission Yeast Schizosaccharomyces pombe. PLoS ONE 2016, 11, e0151286. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Arai, R.; Yashiroda, Y.; Shirai, A.; Kamata, A.; Sekido, S.; Kobayashi, Y.; Hashimoto, A.; Hamamoto, M.; Hiraoka, Y.; et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2006, 24, 841–847. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains/Plasmids | Genotype and Characteristics | Source or Reference |
|---|---|---|
| Fission yeast strains | ||
| 972 | Wild type, h−; the original strain | [20] |
| 975 | Wild type, h+; isogenic to 972 | |
| 968 | Wild type, h90; isogenic to 972 | |
| SP223 | Wild type, h−, ade6–216, leu1–32, ura4–294 | Laboratory collection |
| Plasmids | ||
| pYZx1N Vector Series | ||
| pYZ1N | Fission yeast expression vector with an inducible nmt1 promoter and a LEU2 selectable marker; a derivative of pREP1; wild-type nmt1 promoter with a high level of expression | [11,13] |
| pYZ1N-bleMX6 | Fission yeast expression vector with an inducible nmt1 promoter and a bleMX6 selectable marker; a derivative of pREP1; wild-type nmt1 promoter with a high level of expression | [21] |
| pYZ41N | Same as pYZ1N, but with an intermediate-strength nmt41 promoter; intermediate level of expression | [11,13] |
| pYZ81N | Same as pYZ1N, but with a low-strength nmt81 promoter; low level of expression | [11,13] |
| pYZ2N | Same as pYZ1N but with a URA4 selectable marker | [11,13] |
| pYZ3N-xFP Series | ||
| pYZ3N-GFP | Same as pYZ1N but with a 5′ GFP fluorescent tag | [11] |
| pYZ3N-YFP | Same as pYZ1N but with a 5′ YFP fluorescent tag | Laboratory collection |
| pYZ3N-CFP | Same as pYZ1N but with a 5′ CFP fluorescent tag | Laboratory collection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Benko, Z.; Zhang, C.; Zhao, R.Y. Advanced Protocol for Molecular Characterization of Viral Genome in Fission Yeast (Schizosaccharomyces pombe). Pathogens 2024, 13, 566. https://doi.org/10.3390/pathogens13070566
Zhang J, Benko Z, Zhang C, Zhao RY. Advanced Protocol for Molecular Characterization of Viral Genome in Fission Yeast (Schizosaccharomyces pombe). Pathogens. 2024; 13(7):566. https://doi.org/10.3390/pathogens13070566
Chicago/Turabian StyleZhang, Jiantao, Zsigmond Benko, Chenyu Zhang, and Richard Y. Zhao. 2024. "Advanced Protocol for Molecular Characterization of Viral Genome in Fission Yeast (Schizosaccharomyces pombe)" Pathogens 13, no. 7: 566. https://doi.org/10.3390/pathogens13070566
APA StyleZhang, J., Benko, Z., Zhang, C., & Zhao, R. Y. (2024). Advanced Protocol for Molecular Characterization of Viral Genome in Fission Yeast (Schizosaccharomyces pombe). Pathogens, 13(7), 566. https://doi.org/10.3390/pathogens13070566