
Contributions of Long-Read Sequencing for the Detection of Antimicrobial Resistance

 , , and
, , and
Abstract
:1. Introduction
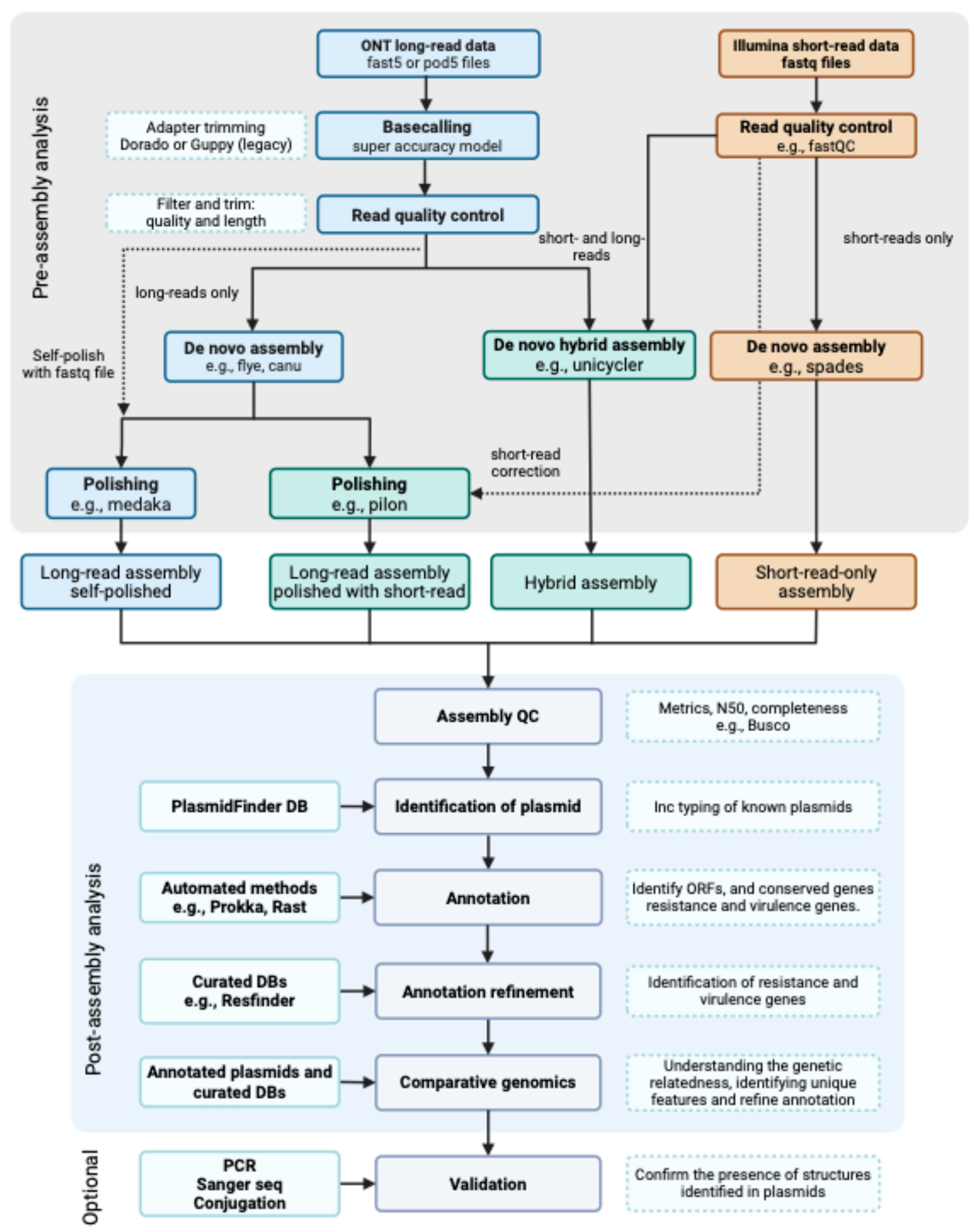
2. Methods
2.1. Bacterial Isolates, Identification, and Carbapenemase Detection
2.2. DNA Extractions and Sequencing
2.3. WGS Data Analysis and Typing
2.4. Conjugations Assays
2.5. Validation of blaNDM Alleles
3. Results
3.1. Sequencing Output
3.2. Assembly, β-Lactamase Identification and Typing
3.3. blaNDM Alleles Confirmation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jauneikaite, E.; Baker, K.S.; Nunn, J.G.; Midega, J.T.; Hsu, L.Y.; Singh, S.R.; Halpin, A.L.; Hopkins, K.L.; Price, J.R.; Srikantiah, P.; et al. Genomics for antimicrobial resistance surveillance to support infection prevention and control in health-care facilities. Lancet Microbe 2023, 4, e1040–e1046. [Google Scholar] [CrossRef] [PubMed]
- Acman, M.; Wang, R.; van Dorp, L.; Shaw, L.P.; Wang, Q.; Luhmann, N.; Yin, Y.; Sun, S.; Chen, H.; Wang, H.; et al. Role of mobile genetic elements in the global dissemination of the carbapenem resistance gene bla(NDM). Nat. Commun. 2022, 13, 1131. [Google Scholar] [CrossRef]
- Wheeler, N.E.; Price, V.; Cunningham-Oakes, E.; Tsang, K.K.; Nunn, J.G.; Midega, J.T.; Anjum, M.F.; Wade, M.J.; Feasey, N.A.; Peacock, S.J.; et al. Innovations in genomic antimicrobial resistance surveillance. Lancet Microbe 2023, 4, e1063–e1070. [Google Scholar] [CrossRef]
- Zhou, X.; Pan, J.; Wang, Y.; Lynch, M.; Long, H.; Zhang, Y. De Novo Structural Variations of Escherichia coli Detected by Nanopore Long-Read Sequencing. Genome Biol. Evol. 2023, 15, evad106. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.; Sierra, R.; Raro, O.H.F.; Aires-de-Sousa, M.; Andrey, D.O.; Nordmann, P. Plasmid-mediated fosfomycin resistance in Escherichia coli isolates of worldwide origin. J. Glob. Antimicrob. Resist. 2023, 35, 137–142. [Google Scholar] [CrossRef]
- Foster-Nyarko, E.; Cottingham, H.; Wick, R.R.; Judd, L.M.; Lam, M.M.C.; Wyres, K.L.; Stanton, T.D.; Tsang, K.K.; David, S.; Aanensen, D.M.; et al. Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, Klebsiella pneumoniae. Microb. Genom. 2023, 9, 000936. [Google Scholar] [CrossRef]
- Bogaerts, B.; Van den Bossche, A.; Verhaegen, B.; Delbrassinne, L.; Mattheus, W.; Nouws, S.; Godfroid, M.; Hoffman, S.; Roosens, N.H.C.; De Keersmaecker, S.C.J.; et al. Closing the gap: Oxford Nanopore Technologies R10 sequencing allows comparable results to Illumina sequencing for SNP-based outbreak investigation of bacterial pathogens. J. Clin. Microbiol. 2024, 62, e0157623. [Google Scholar] [CrossRef] [PubMed]
- Bouras, G.; Judd, L.M.; Edwards, R.A.; Vreugde, S.; Stinear, T.P.; Wick, R.R. How low can you go? Short-read polishing of Oxford Nanopore bacterial genome assemblies. Microb. Genom. 2024, 10, 001254. [Google Scholar] [CrossRef]
- Hall, M.B.; Wick, R.R.; Judd, L.M.; Nguyen, A.N.T.; Steinig, E.J.; Xie, O.; Davies, M.R.; Seemann, T.; Stinear, T.P.; Coin, L.J.M. Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data. eLife 2024, 13. [Google Scholar] [CrossRef]
- Wick, R.R.; Holt, K.E. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research 2019, 8, 2138. [Google Scholar] [CrossRef] [PubMed]
- Schafer, L.; Jehle, J.A.; Kleespies, R.G.; Wennmann, J.T. A practical guide and Galaxy workflow to avoid inter-plasmidic repeat collapse and false gene loss in Unicycler’s hybrid assemblies. Microb. Genom. 2024, 10, 001173. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [CrossRef]
- Dong, H.; Li, Y.; Cheng, J.; Xia, Z.; Liu, W.; Yan, T.; Chen, F.; Wang, Z.; Li, R.; Shi, J.; et al. Genomic Epidemiology Insights on NDM-Producing Pathogens Revealed the Pivotal Role of Plasmids on bla(NDM) Transmission. Microbiol. Spectr. 2022, 10, e0215621. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, X.; Qin, J.; Liang, W.; Shen, Z. Dissemination and Stability of the bla(NDM-5)-Carrying IncX3-Type Plasmid among Multiclonal Klebsiella pneumoniae Isolates. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Creed, T.B.; Reardon, C.L.; Manter, D.K. Comparison of Oxford Nanopore Technologies and Illumina MiSeq sequencing with mock communities and agricultural soil. Sci. Rep. 2023, 13, 9323. [Google Scholar] [CrossRef]
- Watson, M.; Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 2019, 37, 124–126. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Cuber, P.; Chooneea, D.; Geeves, C.; Salatino, S.; Creedy, T.J.; Griffin, C.; Sivess, L.; Barnes, I.; Price, B.; Misra, R. Comparing the accuracy and efficiency of third generation sequencing technologies, Oxford Nanopore Technologies, and Pacific Biosciences, for DNA barcode sequencing applications. Ecol. Genet. Gen. 2023, 28, 100181. [Google Scholar] [CrossRef]
- Li, P.; Luo, W.Y.; Xiang, T.X.; Peng, T.X.; Luo, S.; He, Z.Y.; Liao, W.; Wei, D.D.; Liu, P.; Wan, L.G.; et al. Isolation of Hv-CRKP with co-production of three carbapenemases (bla(KPC), bla(OXA-181) or (OXA-232), and bla(NDM-1)) and a virulence plasmid: A study from a Chinese tertiary hospital. Front. Microbiol. 2023, 14, 1182870. [Google Scholar] [CrossRef]
- Lorenzin, G.; Gona, F.; Battaglia, S.; Spitaleri, A.; Saluzzo, F.; Trovato, A.; di Marco, F.; Cichero, P.; Biancardi, A.; Nizzero, P.; et al. Detection of NDM-1/5 and OXA-48 co-producing extensively drug-resistant hypervirulent in Klebsiella pneumoniae Northern Italy. J. Glob. Antimicrob. Resist. 2022, 28, 146–150. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, X.; Wu, C.; Zhou, P.; Yang, Y.; Wang, B.; Xu, Y.; Zhao, H.; Guo, Y.; Yu, J.; et al. Emergence of KPC-2 and NDM-5-coproducing hypervirulent carbapenem-resistant Klebsiella pneumoniae with high-risk sequence types ST11 and ST15. mSphere 2024, 9, e0061223. [Google Scholar] [CrossRef] [PubMed]
- Sherchan, J.B.; Tada, T.; Shrestha, S.; Uchida, H.; Hishinuma, T.; Morioka, S.; Shahi, R.K.; Bhandari, S.; Twi, R.T.; Kirikae, T.; et al. Emergence of clinical isolates of highly carbapenem-resistant Klebsiella pneumoniae co-harboring blaNDM-5 and blaOXA-181 or -232 in Nepal. J. Infect. Dis. 2020, 92, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Le Terrier, C.; Viguier, C.; Nordmann, P.; Vila, A.J.; Poirel, L.; Carattoli, A. Relative inhibitory activities of the broad-spectrum β-lactamase inhibitor taniborbactam against metallo-β-lactamases. Antimicrob. Agents Chemother. 2024, 68, e0099123. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Akeda, Y.; Takeuchi, D.; Sakamoto, N.; Sugawara, Y.; Yamamoto, N.; Kerdsin, A.; Matsumoto, Y.; Motooka, D.; Leolerd, W.; et al. Clonal dissemination of carbapenem-resistant Klebsiella pneumoniae ST16 co-producing NDM-1 and OXA-232 in Thailand. JAC Antimicrob. Resist. 2022, 4, dlac084. [Google Scholar] [CrossRef]
- Andrey, D.O.; Pereira Dantas, P.; Martins, W.B.S.; Marques De Carvalho, F.; Almeida, L.G.P.; Sands, K.; Portal, E.; Sauser, J.; Cayô, R.; Nicolas, M.F.; et al. An Emerging Clone, Klebsiellapneumoniae Carbapenemase 2–Producing K. pneumoniae Sequence Type 16, Associated With High Mortality Rates in a CC258-Endemic Setting. Clin. Infect. Dis. 2020, 71, e141–e150. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, R.; Liang, Q.; Liang, S.; Li, Q.; Bai, J.; Ding, M.; Zhang, F.; Zhou, Y. Comparison of Two Distinct Subpopulations of Klebsiella pneumoniae ST16 Co-Occurring in a Single Patient. Microbiol. Spectr. 2022, 10, e0262421. [Google Scholar] [CrossRef]
- Hawken, S.E.; Snitkin, E.S. Genomic epidemiology of multidrug-resistant Gram-negative organisms. Ann. N. Y. Acad. Sci. 2019, 1435, 39–56. [Google Scholar] [CrossRef]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Data Set | num_read | mean_len | min_len | med_len | max_len | N50 | total_len | ambiguous_bp | Coverage | avg_read_quality |
|---|---|---|---|---|---|---|---|---|---|---|
| Illumina R1 | 6,451,108 | 100 | 100 | 100 | 100 | 100 | 645,110,800 | 5704 | 234.59 | 39.31 |
| Illumina R2 | 6,451,108 | 100 | 100 | 100 | 100 | 100 | 645,110,800 | 70,826 | * | 38.15 |
| ONT | 630,578 | 3047.7 | 100 | 1299 | 232,397 | 6802 | 1,921,804,262 | 0 | 349.42 | 29.61 |
| Assembly | num_contig | total_assembly_length | max_contig_len | min_contig_len | num_ambig_bp | N50 |
|---|---|---|---|---|---|---|
| Illumina_spades_contigs | 277 | 5,762,046 | 325,640 | 56 | 0 | 152,157 |
| Illumina_spades_scaffolds | 250 | 5,764,032 | 658,511 | 56 | 2610 | 235,693 |
| ONT_flye | 5 | 5,850,368 | 5,371,556 | 5250 | 0 | 5,371,556 |
| ONT_flye_medaka | 5 | 5,850,437 | 5,371,614 | 5253 | 0 | 5,371,614 |
| ONT_flye_pilon | 5 | 5,850,448 | 5,371,627 | 5251 | 0 | 5,371,627 |
| Unicycler (hybrid) | 6 | 5,853,076 | 5,371,783 | 2440 | 0 | 5,371,783 |
| Assembly | Col (pHAD28) | Col440II | IncFIB (pNDM-Mar) | IncHI1B (pNDM-MAR) | IncFII | IncX3 |
|---|---|---|---|---|---|---|
| Illumina_spades | contig 61 (92.25) | contig 47 (100) | contig 27 (99.54) | contig 22 (99.47) | contig 26 (100) | contig 29 (100) |
| Unicycler | contig 6 (92.25) | contig 5 (100) | contig 2 (99.54) | contig 2 (99.47) | contig 3 (100) | contig 4 (100) |
| ONT_flye | contig 6 (92.37) | contig 6 (100) | contig 3 (99.54) | contig 3 (99.47) | contig 4 (100) | contig 1 (100) |
| ONT_flye_medaka | contig 6 (92.37) | contig 6 (100) | contig 3 (99.54) | contig 3 (99.47) | contig 4 (100) | contig 1 (100) |
| ONT_flye_pilon | contig 6 (92.37) | contig 6 (100) | contig 3 (99.54) | contig 3 (99.47) | contig 4 (100) | contig 1 (100) |
| Assembly | MLST | Resistance Gene † | Identity (%) | Contig | Size (bp) | Coverage | Circular | Location § |
|---|---|---|---|---|---|---|---|---|
| Illumina_spades | ST16 | blaNDM-4 | 100 | contig 57 | 3036 | 295 | no | n.d. |
| blaSHV-26 * | 99.88 | contig 1 | 658,511 | 79 | no | chromosome | ||
| blaTEM-1B | 100 | contig 39 | 8779 | 218 | no | n.d. | ||
| Unicycler_hybrid | ST16 | blaNDM-1 | 100 | contig 2 | 352,331 | 1.37× ^ | yes | IncFIB, IncHI1B |
| blaNDM-5 | 100 | contig 4 | 45,181 | 1.42× ^ | yes | IncX3 | ||
| blaSHV-26 * | 99.88 | contig 1 | 5,371,783 | 1.00× ^ | yes | chromosome | ||
| blaTEM-1B | 100 | contig 3 | 76,090 | 1.56× ^ | yes | IncFII | ||
| ONT_flye | ST16 | blaNDM-1 | 100 | contig 3 | 352,292 | 468 | yes | IncFIB, IncHI1B |
| blaNDM-5 | 100 | contig 1 | 45,181 | 844 | yes | IncX3 | ||
| blaSHV-26 * | 99.88 | contig 2 | 5,371,556 | 283 | yes | chromosome | ||
| blaTEM-1B | 100 | contig 4 | 76,089 | 864 | yes | IncFII | ||
| ONT_flye_medaka | ST16 | blaNDM-1 | 100 | contig 3 | 352,300 | 468 | yes | IncFIB, IncHI1B |
| blaNDM-5 | 100 | contig 1 | 45,181 | 844 | yes | IncX3 | ||
| blaSHV-26 * | 99.88 | contig 2 | 5,371,614 | 283 | yes | chromosome | ||
| blaTEM-1B | 100 | contig 4 | 76,089 | 864 | yes | IncFII | ||
| ONT_flye_pilon | ST16 | blaNDM-1 | 100 | contig 3 | 352,300 | 468 | yes | IncFIB, IncHI1B |
| blaNDM-5 | 100 | contig 1 | 45,181 | 844 | yes | IncX3 | ||
| blaSHV-26 * | 99.88 | contig 2 | 5,371,627 | 283 | yes | chromosome | ||
| blaTEM-1B | 100 | contig 4 | 76,089 | 864 | yes | IncFII |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sierra, R.; Roch, M.; Moraz, M.; Prados, J.; Vuilleumier, N.; Emonet, S.; Andrey, D.O. Contributions of Long-Read Sequencing for the Detection of Antimicrobial Resistance. Pathogens 2024, 13, 730. https://doi.org/10.3390/pathogens13090730
Sierra R, Roch M, Moraz M, Prados J, Vuilleumier N, Emonet S, Andrey DO. Contributions of Long-Read Sequencing for the Detection of Antimicrobial Resistance. Pathogens. 2024; 13(9):730. https://doi.org/10.3390/pathogens13090730
Chicago/Turabian StyleSierra, Roberto, Mélanie Roch, Milo Moraz, Julien Prados, Nicolas Vuilleumier, Stéphane Emonet, and Diego O. Andrey. 2024. "Contributions of Long-Read Sequencing for the Detection of Antimicrobial Resistance" Pathogens 13, no. 9: 730. https://doi.org/10.3390/pathogens13090730