
Gammaherpesvirus Infection Stimulates Lung Tumor-Promoting Inflammation
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. MHV68 Preparation
2.2. Animal Model
2.3. RNA Preparation and Analysis
2.4. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
2.5. Protein Extraction of Mouse Lung Tissue
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Tumor Counting
2.8. Immunofluorescence
2.9. Flow Cytometry
2.10. Immunoprecipitation of Methylated mRNAs (m6A Me-RIP Assay)
2.11. Statistical Analysis
3. Results
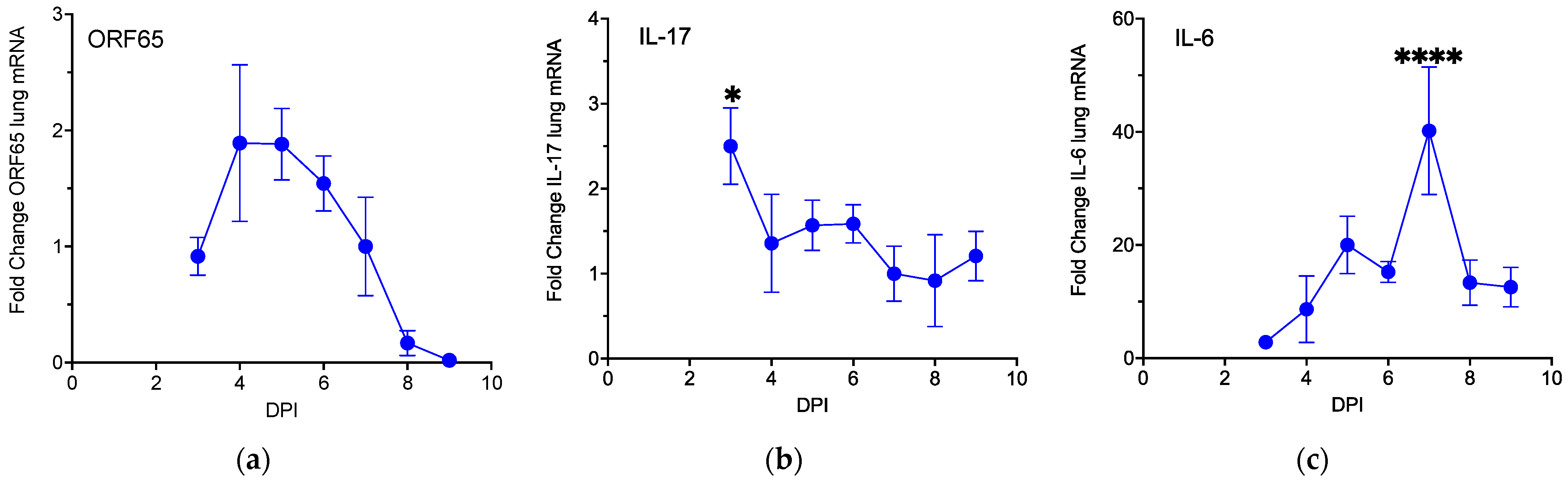
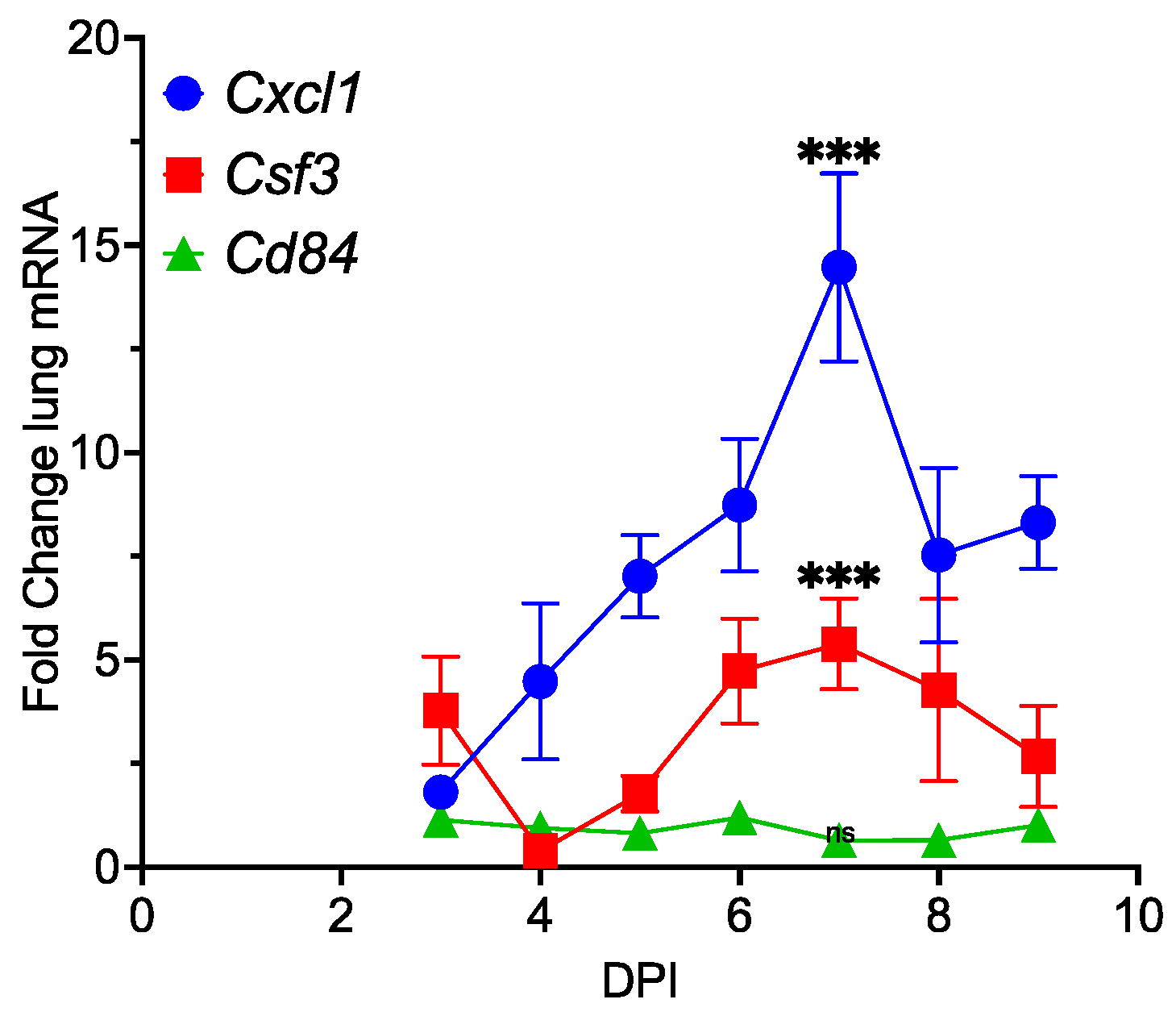
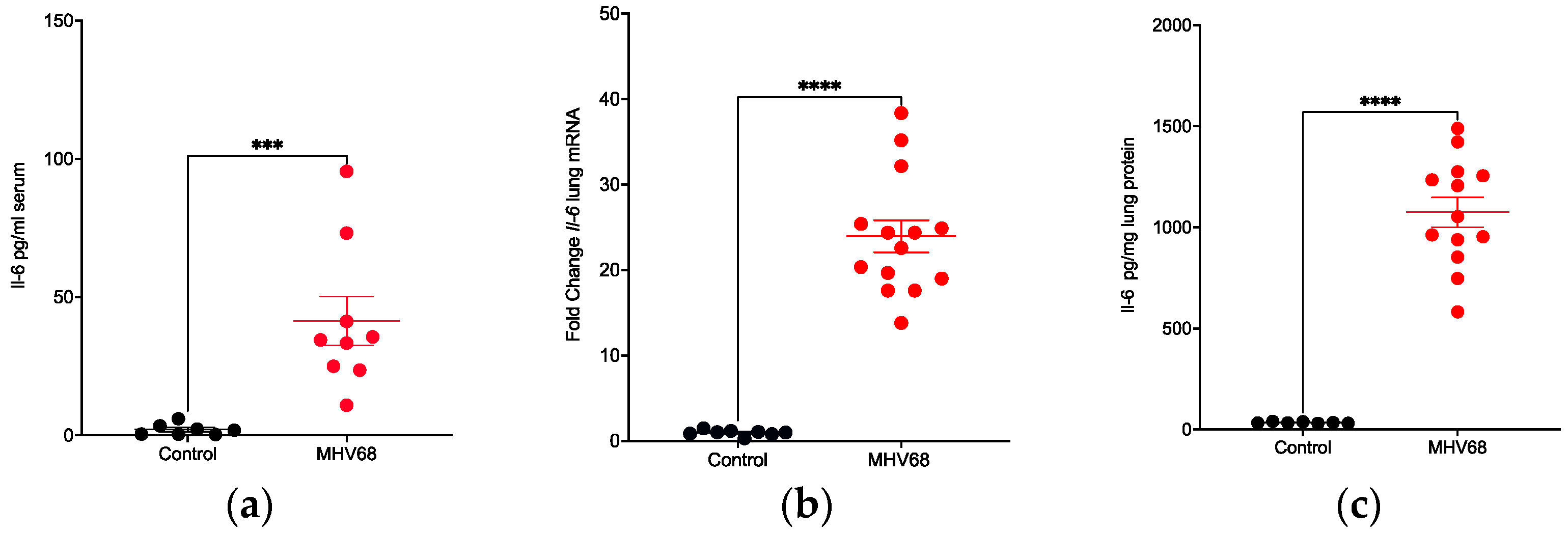
3.1. MHV68 Induces Type 17 Cytokines during Virus Infection
3.2. MHV68 Infection Promotes Proliferative Lesions in Tumor-Bearing Mice
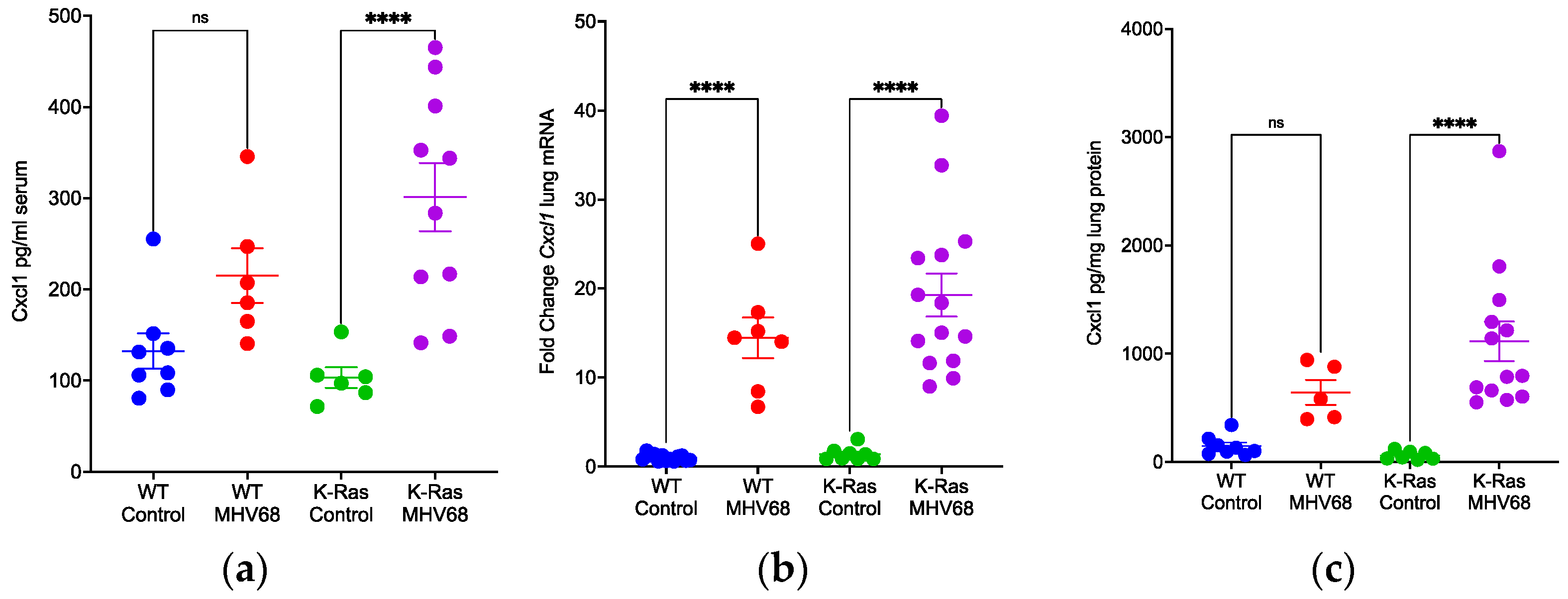
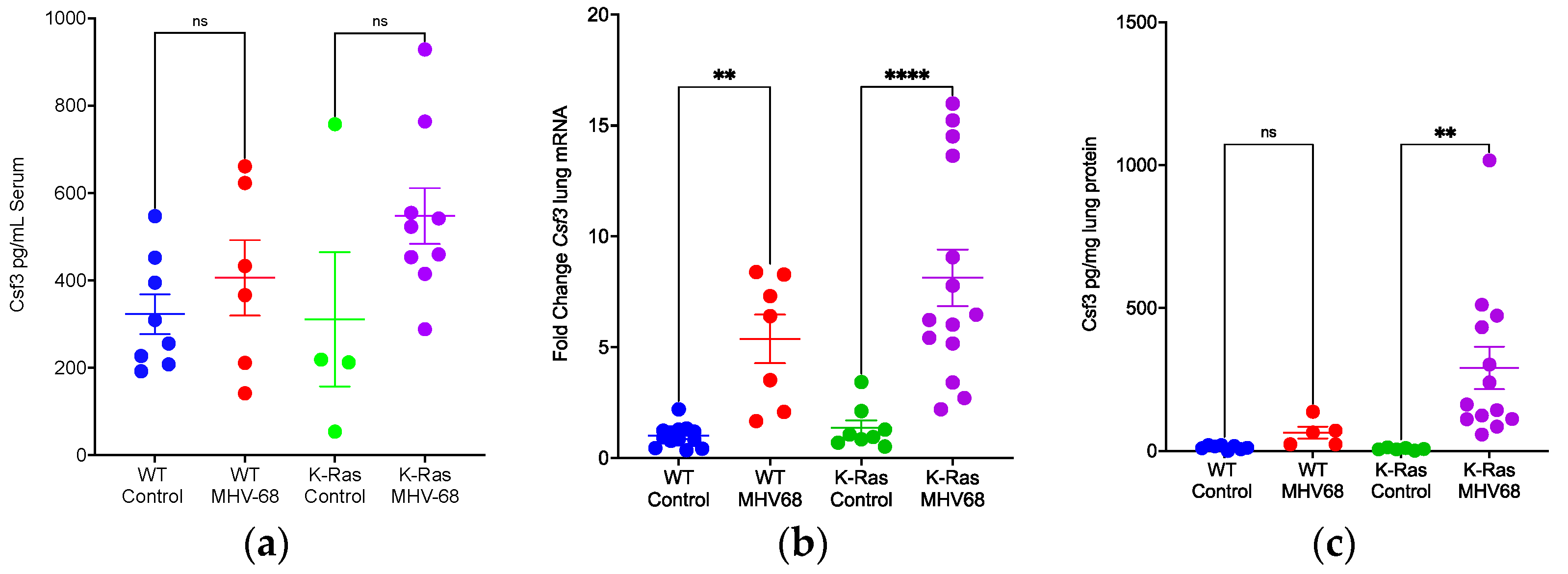
3.3. Promotion of Inflammation by MHV68 Infection
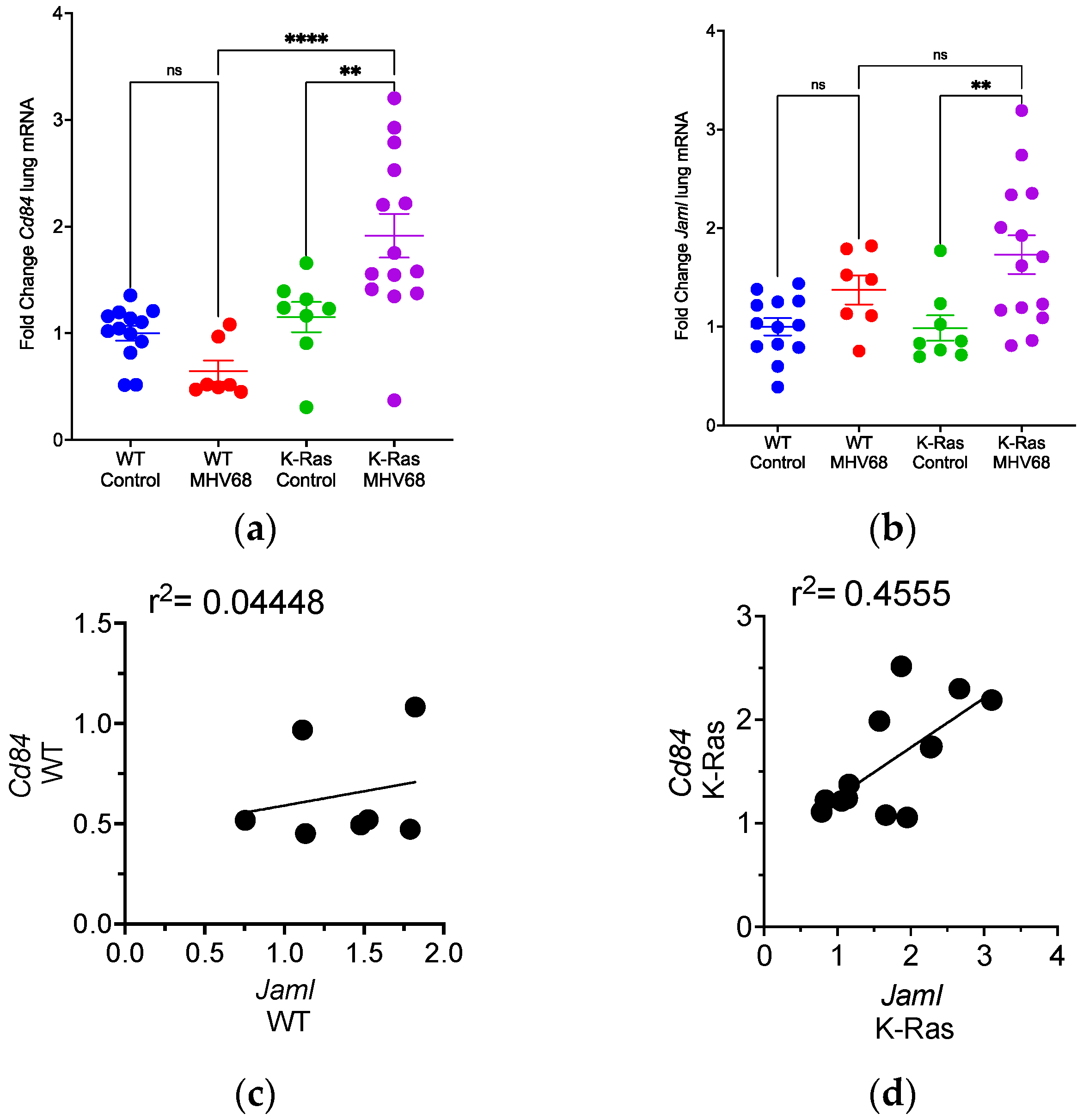
3.4. Disparate Recruitment of Immune Cells in Wild-Type and K-RasLA1 Mice
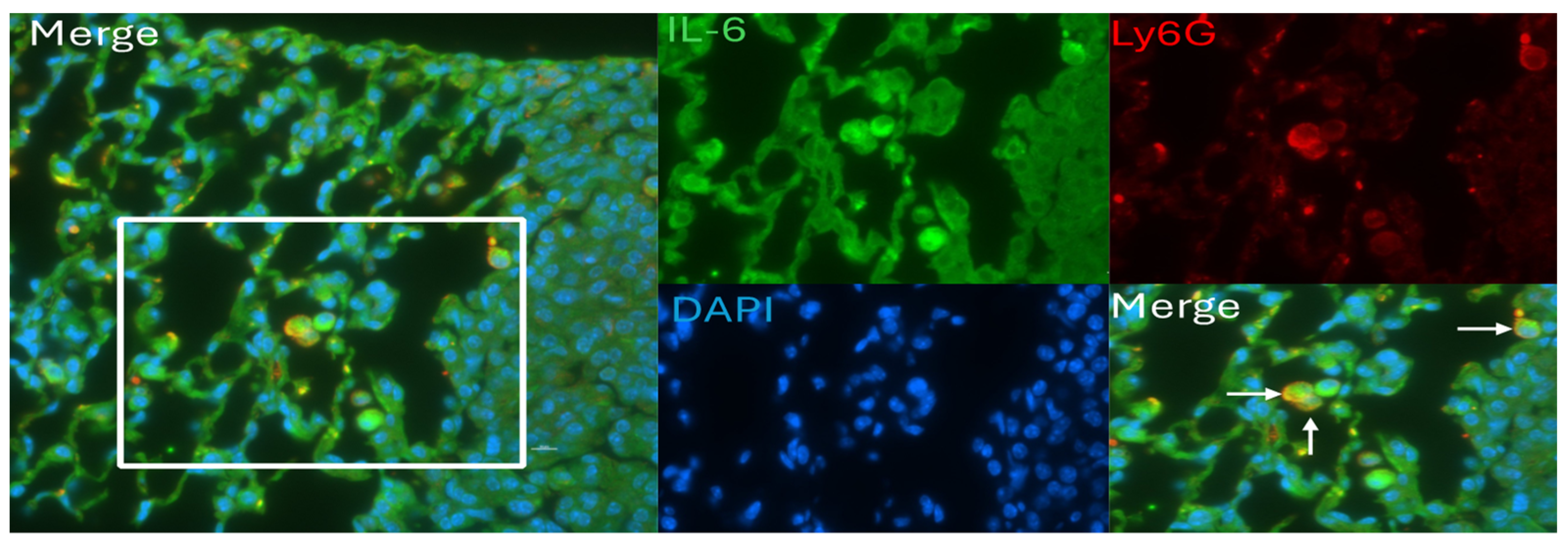
3.5. IL-6 Induction and Localization in Tumor-Bearing Mice
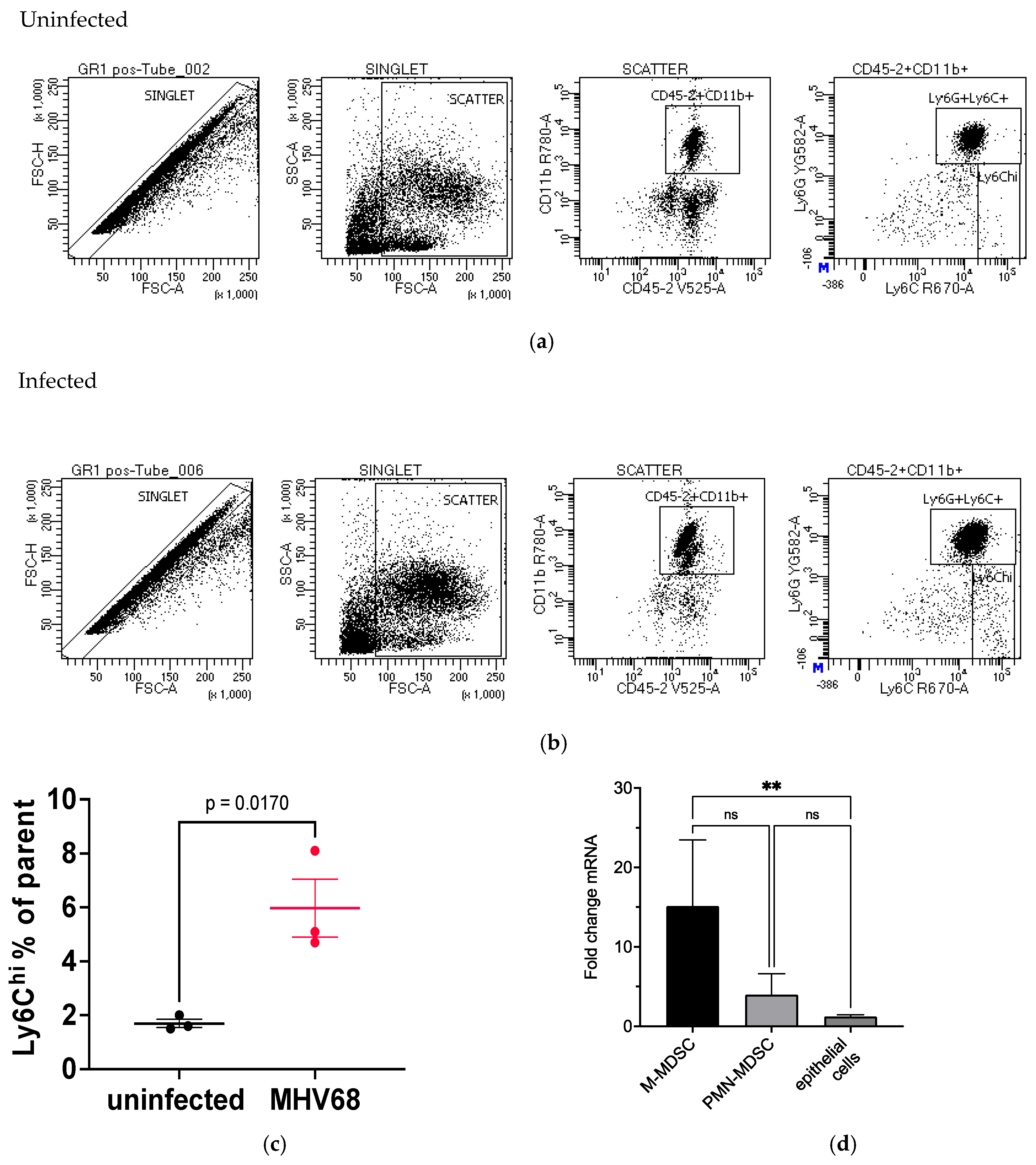
3.6. Evaluation of Immune Cells in MHV68-Infected Tumor-Bearing Mice
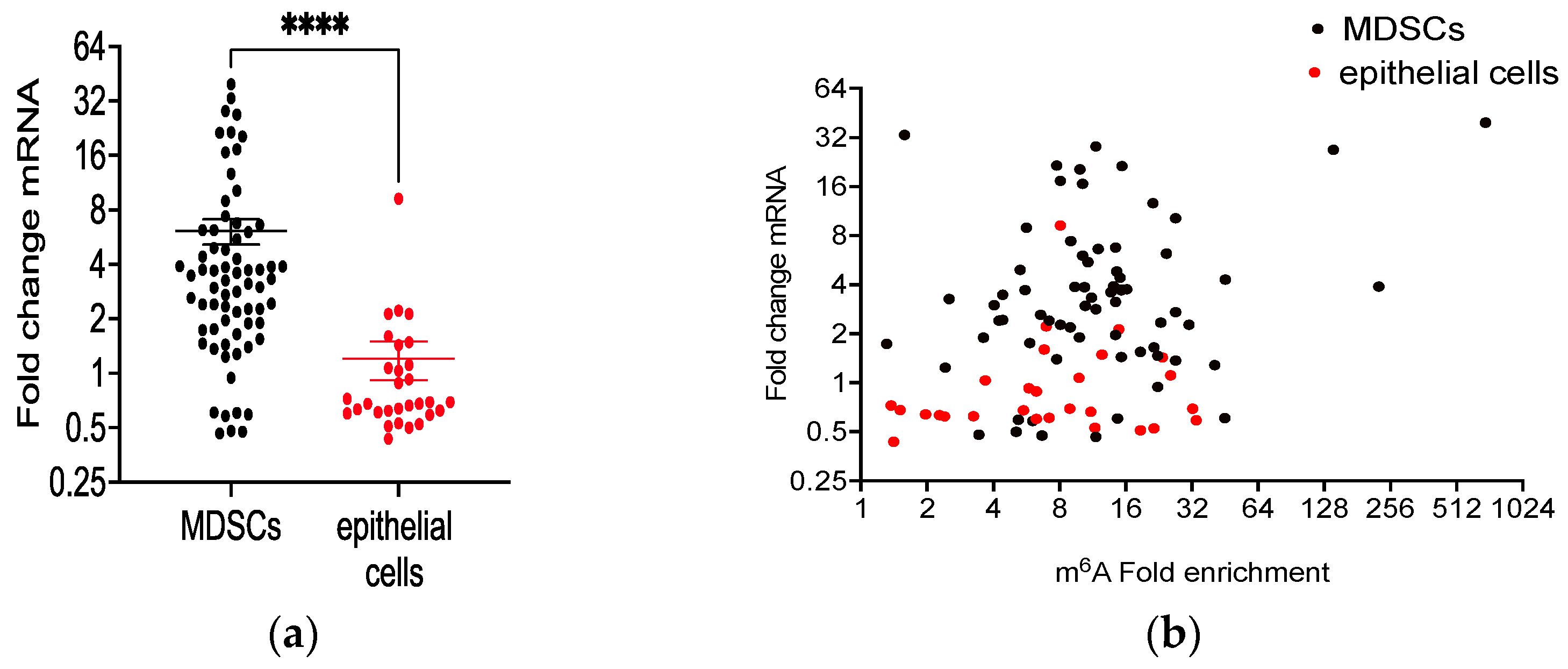
3.7. Differential m6A Methylation of Immune Cell Selective mRNAs in MHV68-Infected Tumor-Bearing Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Loeb, L.A.; Ernster, V.L.; Warner, K.E.; Abbotts, J.; Laszlo, J. Smoking and lung cancer: An overview. Cancer Res. 1984, 44, 5940–5958. [Google Scholar]
- Xu, B.; Guenther, J.F.; Pociask, D.A.; Wang, Y.; Kolls, J.K.; You, Z.; Chandrasekar, B.; Shan, B.; Sullivan, D.E.; Morris, G.F. Promotion of lung tumor growth by interleukin-17. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2014, 307, L497–L508. [Google Scholar] [CrossRef]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023, 23, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.C.; Marongiu, F.; Evans, E.J., Jr.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [CrossRef]
- Yao, Z.; Fanslow, W.C.; Seldin, M.F.; Rousseau, A.M.; Painter, S.L.; Comeau, M.R.; Cohen, J.I.; Spriggs, M.K. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 1995, 3, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Tosato, G. Viral interleukin-6: Role in Kaposi’s sarcoma-associated herpesvirus: Associated malignancies. J. Interferon Cytokine Res. 2011, 31, 791–801. [Google Scholar] [CrossRef]
- Jondle, C.N.; Johnson, K.E.; Aurubin, C.; Sylvester, P.; Xin, G.; Cui, W.; Huppler, A.R.; Tarakanova, V.L. Gammaherpesvirus Usurps Host IL-17 Signaling To Support the Establishment of Chronic Infection. mBio 2021, 12, e00566-21. [Google Scholar] [CrossRef] [PubMed]
- Jondle, C.N.; Tarakanova, V.L. T Cell-Intrinsic Interleukin 17 Receptor A Signaling Supports the Establishment of Chronic Murine Gammaherpesvirus 68 Infection. J. Virol. 2022, 96, e0063922. [Google Scholar] [CrossRef]
- Guenther, J.F.; Cameron, J.E.; Nguyen, H.T.; Wang, Y.; Sullivan, D.E.; Shan, B.; Lasky, J.A.; Flemington, E.K.; Morris, G.F. Modulation of lung inflammation by the Epstein-Barr virus protein Zta. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L771–L784. [Google Scholar] [CrossRef]
- Wang, Y.; Tibbetts, S.A.; Krug, L.T. Conquering the Host: Determinants of Pathogenesis Learned from Murine Gammaherpesvirus 68. Annu. Rev. Virol. 2021, 8, 349–371. [Google Scholar] [CrossRef]
- Liang, X.; Collins, C.M.; Mendel, J.B.; Iwakoshi, N.N.; Speck, S.H. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 2009, 5, e1000677. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein-Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Tzellos, S.; Farrell, P.J. Epstein-barr virus sequence variation-biology and disease. Pathogens 2012, 1, 156–174. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Shechter, O.; Sausen, D.G.; Gallo, E.S.; Dahari, H.; Borenstein, R. Epstein-Barr Virus (EBV) Epithelial Associated Malignancies: Exploring Pathologies and Current Treatments. Int. J. Mol. Sci. 2022, 23, 14389. [Google Scholar] [CrossRef]
- Han, S.; Tay, J.K.; Loh, C.J.L.; Chu, A.J.M.; Yeong, J.P.S.; Lim, C.M.; Toh, H.C. Epstein-Barr Virus Epithelial Cancers-A Comprehensive Understanding to Drive Novel Therapies. Front. Immunol. 2021, 12, 734293. [Google Scholar] [CrossRef]
- Becnel, D.; Abdelghani, R.; Nanbo, A.; Avilala, J.; Kahn, J.; Li, L.; Lin, Z. Pathogenic Role of Epstein-Barr Virus in Lung Cancers. Viruses 2021, 13, 877. [Google Scholar] [CrossRef] [PubMed]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.C.; Blanco, R.; Corvalán, A.H.; Muñoz, J.P.; Calaf, G.M.; Aguayo, F. Epstein-Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens 2022, 11, 132. [Google Scholar] [CrossRef]
- Park, M.D.; Reyes-Torres, I.; LeBerichel, J.; Hamon, P.; LaMarche, N.M.; Hegde, S.; Belabed, M.; Troncoso, L.; Grout, J.A.; Magen, A.; et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat. Immunol. 2023, 24, 792–801. [Google Scholar] [CrossRef]
- Virgin, H.W.t.; Presti, R.M.; Li, X.Y.; Liu, C.; Speck, S.H. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 1999, 73, 2321–2332. [Google Scholar] [CrossRef]
- Gargano, L.M.; Forrest, J.C.; Speck, S.H. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol. 2009, 83, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Usherwood, E.J.; Stewart, J.P.; Nash, A.A.; Sun, R. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 2000, 74, 3659–3667. [Google Scholar] [CrossRef]
- Weck, K.E.; Barkon, M.L.; Yoo, L.I.; Speck, S.H.; Virgin, H.I. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 1996, 70, 6775–6780. [Google Scholar] [CrossRef]
- Raffegerst, S.; Steer, B.; Hohloch, M.; Adler, H. Prevention of Tumor Formation by Latent Gammaherpesvirus Infection. PLoS ONE 2015, 10, e0145678. [Google Scholar] [CrossRef]
- Forrest, J.C.; Speck, S.H. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 2008, 82, 7688–7699. [Google Scholar] [CrossRef]
- Hoffmann, S.H.L.; Reck, D.I.; Maurer, A.; Fehrenbacher, B.; Sceneay, J.E.; Poxleitner, M.; Öz, H.H.; Ehrlichmann, W.; Reischl, G.; Fuchs, K.; et al. Visualization and quantification of in vivo homing kinetics of myeloid-derived suppressor cells in primary and metastatic cancer. Theranostics 2019, 9, 5869–5885. [Google Scholar] [CrossRef] [PubMed]
- Bechara, R.; Amatya, N.; Bailey, R.D.; Li, Y.; Aggor, F.E.Y.; Li, D.D.; Jawale, C.V.; Coleman, B.M.; Dai, N.; Gokhale, N.S.; et al. The m(6)A reader IMP2 directs autoimmune inflammation through an IL-17- and TNFα-dependent C/EBP transcription factor axis. Sci. Immunol. 2021, 6, eabd1287. [Google Scholar] [CrossRef] [PubMed]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, aay6017. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Li, X.; Bechara, R.; Zhao, J.; McGeachy, M.J.; Gaffen, S.L. IL-17 receptor-based signaling and implications for disease. Nat. Immunol. 2019, 20, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Novotny, M.; Bulek, K.; Liu, C.; Li, X.; Hamilton, T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 2011, 12, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e817. [Google Scholar] [CrossRef]
- Ford, J.W.; Gonzalez-Cotto, M.; MacFarlane, A.W.t.; Peri, S.; Howard, O.M.Z.; Subleski, J.J.; Ruth, K.J.; Haseebuddin, M.; Al-Saleem, T.; Yang, Y.; et al. Tumor-Infiltrating Myeloid Cells Co-Express TREM1 and TREM2 and Elevated TREM-1 Associates With Disease Progression in Renal Cell Carcinoma. Front. Oncol. 2021, 11, 662723. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, Y.; Bai, H.; Jiao, S. Systematic assessment of the clinicopathological prognostic significance of tissue cytokine expression for lung adenocarcinoma based on integrative analysis of TCGA data. Sci. Rep. 2019, 9, 6301. [Google Scholar] [CrossRef]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Ibrahim, M.L.; Lu, C.; Klement, J.D.; Redd, P.S.; Yang, D.; Smith, A.D.; Liu, K. Expression profiles and function of IL6 in polymorphonuclear myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2020, 69, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dou, X.; Chen, S.; Yu, X.; Huang, X.; Zhang, L.; Chen, Y.; Wang, J.; Yang, K.; Bugno, J.; et al. YTHDF2 inhibition potentiates radiotherapy antitumor efficacy. Cancer Cell 2023, 41, 1294–308.e1298. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e1016. [Google Scholar] [CrossRef]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell. Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Stathopoulos, G.P.; Armakolas, A.; Tranga, T.; Marinou, H.; Stathopoulos, J.; Chandrinou, H. Granulocyte colony-stimulating factor expression as a prognostic biomarker in non-small cell lung cancer. Oncol. Rep. 2011, 25, 1541–1544. [Google Scholar] [CrossRef]
- Ganesan, R.; Bhasin, S.S.; Bakhtiary, M.; Krishnan, U.; Cheemarla, N.R.; Thomas, B.E.; Bhasin, M.K.; Sukhatme, V.P. Taxane chemotherapy induces stromal injury that leads to breast cancer dormancy escape. PLoS Biol. 2023, 21, e3002275. [Google Scholar] [CrossRef]
- Rodrigues, C.F.D.; Serrano, E.; Patrício, M.I.; Val, M.M.; Albuquerque, P.; Fonseca, J.; Gomes, C.M.F.; Abrunhosa, A.J.; Paiva, A.; Carvalho, L.; et al. Stroma-derived IL-6, G-CSF and Activin-A mediated dedifferentiation of lung carcinoma cells into cancer stem cells. Sci. Rep. 2018, 8, 11573. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Wei, J.J.; Yuan, Y.; Sun, R.; Li, D.; Luo, J.; Liao, S.J.; Zhou, Y.H.; Shu, Y.; Wang, Q.; et al. IL-6 cooperates with G-CSF to induce protumor function of neutrophils in bone marrow by enhancing STAT3 activation. J. Immunol. 2013, 190, 5882–5893. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Juan, C.Y.; Chang, K.J.; Chen, W.J.; Kuo, M.L. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis 2001, 22, 1947–1953. [Google Scholar] [CrossRef]
- Jourdan, M.; Veyrune, J.L.; De Vos, J.; Redal, N.; Couderc, G.; Klein, B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene 2003, 22, 2950–2959. [Google Scholar] [CrossRef]
- Wegiel, B.; Bjartell, A.; Culig, Z.; Persson, J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 2008, 122, 1521–1529. [Google Scholar] [CrossRef]
- Zou, M.; Zhang, X.; Xu, C. IL6-induced metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of 3,3’-diindolylmethane in ovarian cancer cells. Cell Oncol. 2016, 39, 47–57. [Google Scholar] [CrossRef]
- Barrio, L.; Gaspar, A.E.; Muzzopappa, M.; Ghosh, K.; Romao, D.; Clemente-Ruiz, M.; Milán, M. Chromosomal instability-induced cell invasion through caspase-driven DNA damage. Curr. Biol. 2023, 33, 4446–4457.e5. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Jerman, S.J.; Jacenik, D.; Phinney, B.B.; Yao, R.; Prossnitz, E.R.; Beswick, E.J. G-CSF and G-CSFR Modulate CD4 and CD8 T Cell Responses to Promote Colon Tumor Growth and Are Potential Therapeutic Targets. Front. Immunol. 2020, 11, 1885. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Li, X.; Guo, X.; Lu, Y.; Xie, Y.; Huang, X.; Lin, J.; Tan, W.; Wang, C. Myeloid-derived suppressor cells promote tumor growth and sorafenib resistance by inducing FGF1 upregulation and fibrosis. Neoplasia 2022, 28, 100788. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward Sequence | Reverse Sequence |
|---|---|---|
| ORF65 | 5′GTCAGGGCCCAGTCCGTA3′ | 5′TGGCCCTCTACCTTCTGTTGA3′ |
| IL-17A | 5′CAGACTACCTCAACCGTTCCAC3′ | 5′TCCAGCTTTCCCTCCGCATTGA3′ |
| IL-6 | 5′TACCACTTCACAAGTCGGAGGC3′ | 5′CTGCAAGTGCATCATCGTTGTT3′ |
| CXCL1 | 5′CTGGGATTCACCTCAAGAACATC3′ | 5′CAGGGTCAAGGCAAGCCTC3′ |
| CSF3 | 5′ATGGCTCAACTTTCTGCCCAG3′ | 5′CTGACAGTGACCAGGGGAAC3′ |
| CD84 | 5′ATATAGCTGGAGTCCCTTTGGAG3′ | 5′AAAGAGCACGGCCAATCCTC3′ |
| JAML | 5′ATGCTTTGCCTCCTGAAACTG3′ | 5′TGATTCACCCACATGCACTCT3′ |
| ß-actin | 5′GATGTATGAAAGCTTTGGTC3′ | 5′TGTGCACTTTTATTGGTCTC3′ |
| Antibody | Detection | Source and Catalog Number |
|---|---|---|
| Purified Rat Anti-Mouse Ly6G (1A8) | Ly6G | BD Pharmingen cat#: 551459/clone1A8 |
| Rabbit IL-6 Antibody | IL-6 | Novus Biologicals cat#: NB600-1131 |
| Goat Anti-Rat IgG H&L (Alexa Fluor 647) | Secondary anti-rat | Abcam cat#: ab150159 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor 488) | Secondary anti-rabbit | Abcam cat#: ab150077 |
| Rat APC-eFluor780 CD11b | CD11b | Invitrogen cat#: 47-0112-80/clone M1/70 |
| Rat APC Anti-Mouse Ly6C | Ly6C | BioLegend cat#: 128015/clone HK1.4 |
| Mouse V500 Mouse Anti-Mouse CD45.2 | CD45.2 | BD Horizon cat#: 562130/clone 104 |
| PE Rat Anti-Mouse Ly6G (1A8) | Ly6G | BD Pharmingen/cat#: 561104/clone 1A8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukhopadhyay, S.S.; Swan, K.F.; Pridjian, G.; Kolls, J.K.; Zhuang, Y.; Yin, Q.; Lasky, J.A.; Flemington, E.; Morris, C.A.; Lin, Z.; et al. Gammaherpesvirus Infection Stimulates Lung Tumor-Promoting Inflammation. Pathogens 2024, 13, 747. https://doi.org/10.3390/pathogens13090747
Mukhopadhyay SS, Swan KF, Pridjian G, Kolls JK, Zhuang Y, Yin Q, Lasky JA, Flemington E, Morris CA, Lin Z, et al. Gammaherpesvirus Infection Stimulates Lung Tumor-Promoting Inflammation. Pathogens. 2024; 13(9):747. https://doi.org/10.3390/pathogens13090747
Chicago/Turabian StyleMukhopadhyay, Sudurika S., Kenneth F. Swan, Gabriella Pridjian, Jay K. Kolls, Yan Zhuang, Qinyan Yin, Joseph A. Lasky, Erik Flemington, Cindy A. Morris, Zhen Lin, and et al. 2024. "Gammaherpesvirus Infection Stimulates Lung Tumor-Promoting Inflammation" Pathogens 13, no. 9: 747. https://doi.org/10.3390/pathogens13090747