
Diverse Head-to-Tail Sequences in the Circular Genome of Human Bocavirus Genotype 1 among Children with Acute Respiratory Infections Implied the Switch of Template Chain in the Rolling-Circle Replication Model

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. Nucleic Acid Extraction
2.3. CEMP Assay for Multiple-Pathogen Screening
2.4. Genotyping of HBoVs by PCR
2.5. Amplification, Cloning, and Sanger Sequencing of Head-to-Tail Sequences and Assembly of the HBoV1 Circular Genome
2.6. Amplicon Sequencing of Nested PCR Amplification Products
2.7. Sequence Analysis of the Head-to-Tail Sequence
2.8. Meta-Genomic Next-Generation Sequencing (mNGS) of Circular Genomes
3. Results
3.1. Pathogen Screening by the CEMP Assay
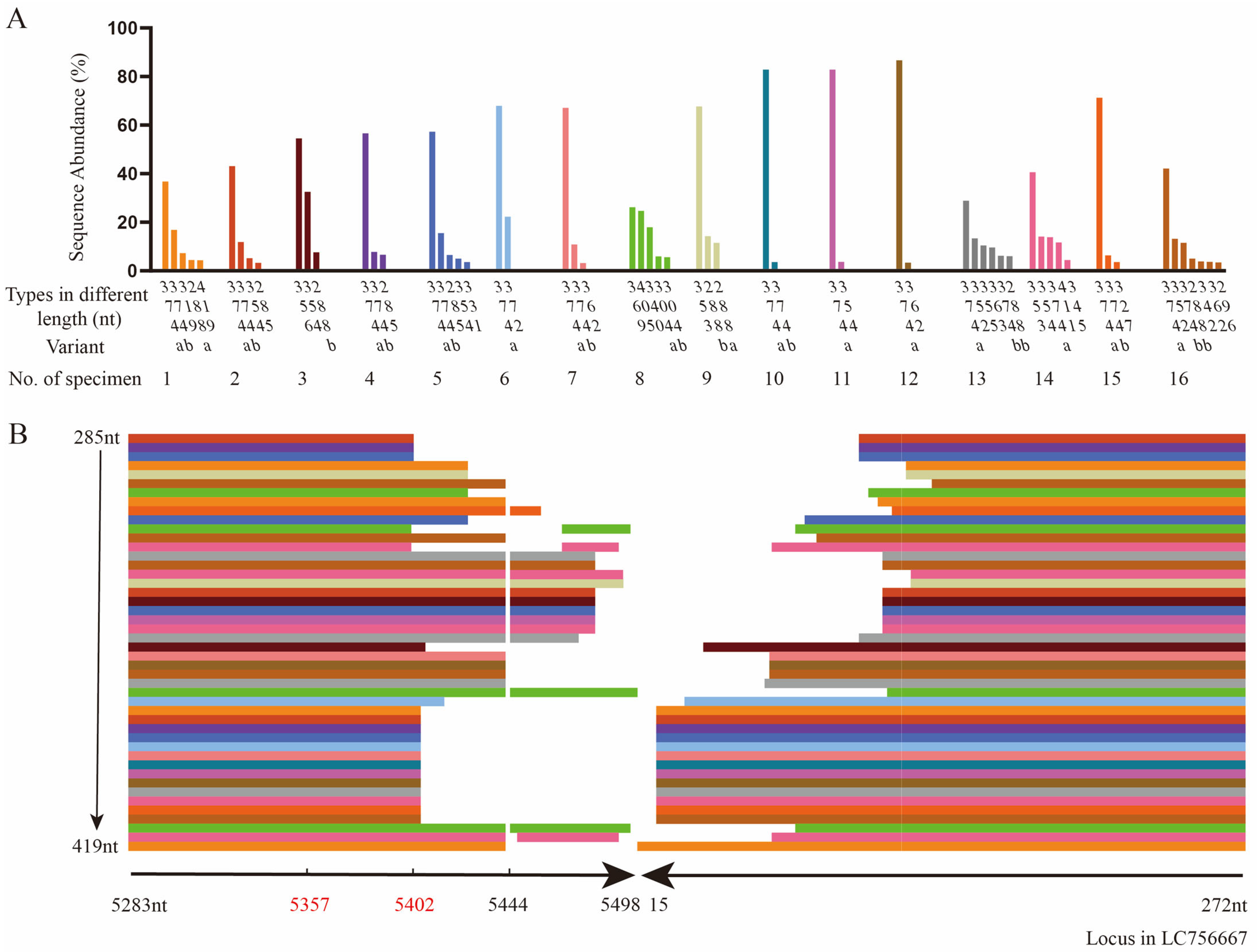
3.2. The Head-to-Tail Sequences of HBoV1 by TA Cloning and Sanger Sequencing
3.3. The Head-to-Tail Sequences of HBoV1 by Amplicon Sequencing
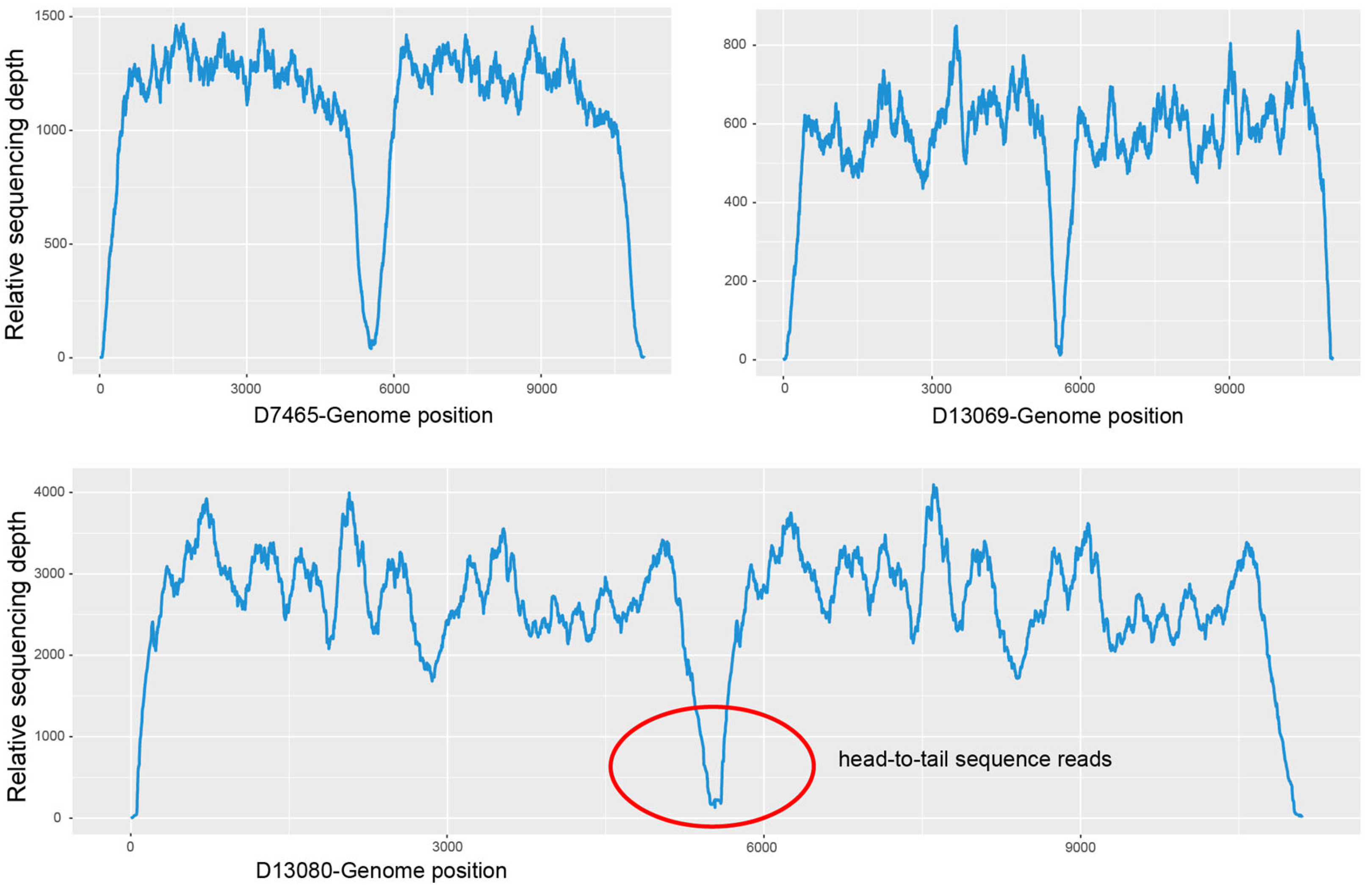
3.4. Meta-Genomic Next-Generation Sequencing (mNGS)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 15712. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Slikas, E.; Simmonds, P.; Chieochansin, T.; Naeem, A.; Shaukat, S.; Alam, M.M.; Sharif, S.; Angez, M.; Zaidi, S.; et al. A newly identified bocavirus species in human stool. J. Infect. Dis. 2009, 199, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Slikas, E.; Li, L.; Bodhidatta, L.; Sethabutr, O.; Triki, H.; Bahri, O.; Oderinde, B.S.; Baba, M.M.; et al. Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J. Infect. Dis. 2010, 201, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.L.; Higgins, G.D.; Davidson, G.P.; Givney, R.C.; Ratcliff, R.M. A novel bocavirus associated with acute gastroenteritis in Australian children. PLoS Pathog. 2009, 5, 11. [Google Scholar] [CrossRef]
- De, R.; Zhang, K.-X.; Wang, F.; Zhou, Y.-T.; Sun, Y.; Chen, D.-M.; Zhu, R.-N.; Guo, Q.; Liu, S.; Qu, D.; et al. Human bocavirus 1 is a genuine pathogen for acute respiratory tract infection in pediatric patients determined by nucleic acid, antigen, and serology tests. Front. Microbiol. 2022, 13, 9. [Google Scholar] [CrossRef]
- Christensen, A.; Kesti, O.; Elenius, V.; Eskola, A.L.; Døllner, H.; Altunbulakli, C.; Akdis, C.A.; Söderlund-Venermo, M.; Jartti, T. Human bocaviruses and pediatric infections. Lancet Child. Adolesc. Health 2019, 3, 418–426. [Google Scholar] [CrossRef]
- Rajme-López, S. Human Bocavirus-1 infection: A closer look into its clinical and virological features. Lancet Reg. Health Am. 2024, 29, 100672. [Google Scholar] [CrossRef]
- Trapani, S.; Caporizzi, A.; Ricci, S.; Indolfi, G. Human Bocavirus in Childhood: A True Respiratory Pathogen or a “Passenger” Virus? A Comprehensive Review. Microorganisms 2023, 11, 1243. [Google Scholar] [CrossRef]
- Böhmer, A.; Schildgen, V.; Lüsebrink, J.; Ziegler, S.; Tillmann, R.L.; Kleines, M.; Schildgen, O. Novel application for isothermal nucleic acid sequence-based amplification (NASBA). J. Virol. Methods 2009, 158, 199–201. [Google Scholar] [CrossRef]
- Zhao, L.Q.; Qian, Y.; Zhu, R.N.; Deng, J.; Wang, F. Genomic sequence analysis for human bocavirus circulating in Beijing by bioinformatics. Chin. J. Microbiol. Immunol. 2007, 27, 389–393. [Google Scholar]
- Shen, W.; Deng, X.; Zou, W.; Cheng, F.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Identification and functional analysis of novel nonstructural proteins of human bocavirus 1. J Virol. 2015, 89, 10097–10109. [Google Scholar] [CrossRef]
- Zou, W.; Cheng, F.; Shen, W.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Nonstructural protein NP1 of human bocavirus 1 plays a critical role in the expression of viral capsid proteins. J. Virol. 2016, 90, 4658–4669. [Google Scholar] [CrossRef]
- Huang, Q.; Deng, X.; Yan, Z.; Cheng, F.; Luo, Y.; Shen, W.; Lei-Butters, D.C.M.; Chen, A.Y.; Li, Y.; Tang, L.; et al. Establishment of a reverse genetics system for studying human bocavirus in human airway epithelia. PLoS Pathog. 2012, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Deng, X.; Zou, W.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Analysis of cis and trans requirements for DNA replication at the right-end hairpin of the human bocavirus 1 genome. J. Virol. 2016, 90, 7761–7777. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef]
- Tattersall, P.; Ward, D.C. Rolling hairpin model for replication of parvovirus and linear chromosomal DNA. Nature 1976, 263, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Genome packaging sense is controlled by the efficiency of the nick site in the right-end replication origin of parvoviruses minute virus of mice and LuIII. J. Virol. 2005, 79, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. High-mobility group 1/2 proteins are essential for initiating rolling-circle-type DNA replication at a parvovirus hairpin origin. J. Virol. 1998, 72, 8477–8484. [Google Scholar] [CrossRef]
- Lüsebrink, J.; Schildgen, V.; Tillmann, R.L.; Wittleben, F.; Böhmer, A.; Müller, A.; Schildgen, O. Detection of head-to-tail DNA sequences of human bocavirus in clinical samples. PLoS ONE 2011, 6, e19457. [Google Scholar] [CrossRef]
- Kapoor, A.; Hornig, M.; Asokan, A.; Williams, B.; Henriquez, J.A.; Lipkin, W.I. Bocavirus episome in infected human tissue contains non-identical termini. PLoS ONE 2011, 6, e21362. [Google Scholar] [CrossRef]
- Babkin, I.V.; Tyumentsev, A.I.; Tikunov, A.Y.; Zhirakovskaia, E.V.; Netesov, S.V.; Tikunova, N.V. A study of the human bocavirus replicative genome structures. Virus Res. 2015, 195, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, L.; Sun, Y.; Qian, Y.; Liu, L.; Jia, L.; Zhang, Y.; Dong, H. Detection of a bocavirus circular genome in fecal specimens from children with acute diarrhea in Beijing, China. PLoS ONE 2012, 7, e48980. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, T.; Qian, Y.; Song, J.; Zhu, R.; Liu, L.; Jia, L.; Dong, H. Keeping all secondary structures of the non-coding region in the circular genome of human bocavirus 2 is important for DNA replication and virus assembly, as revealed by three hetero-recombinant genomic clones. Emerg. Microbes Infect. 2019, 8, 1563–1573. [Google Scholar] [CrossRef]
- Doermann, A.H. T4 and the rolling circle model of replication. Annu. Rev. Genet. 1973, 7, 325–341. [Google Scholar] [CrossRef]
- Li, X.; Chen, B.; Zhang, S.; Li, X.; Chang, J.; Tang, Y.; Wu, Y.; Lu, X. Rapid detection of respiratory pathogens for community-acquired pneumonia by capillary electrophoresis-based multiplex PCR. SLAS Technol. 2019, 24, 105–116. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, R.; Qian, Y.; Deng, J.; Wang, F.; Sun, Y.; Dong, H.; Liu, L.; Jia, L.; Zhao, L. Prevalence analysis of different human bocavirus genotypes in pediatric patients revealed intra-genotype recombination. Infect. Genet. Evol. 2014, 27, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Cheung, A.K. Detection of template strand switching during initiation and termination of DNA replication of porcine circovirus. J. Virol. 2004, 78, 4268–4277. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round | Name | Position | Sequences (5′-3′) |
|---|---|---|---|
| First | HBoV1-F1-Tail (+) | JQ923422:5187-5206 | gcttctgcttacaagttcct |
| HBoV1-R1-Head (−) | JQ923422:365-346 | ggaggattgaaagccatagt | |
| Second | HBoV1-F2-Tail (+) | JQ923422:5283-5300 | tggtgttaccgtctcgaa |
| HBoV1-R2-Head (−) | JQ923422:272-254 | aggaagtgcagcagcttaa |
| Type | No. of Sequences | 5′ REH Located on the Position of JQ923422 | Linker Sequences | 3′ LEH Located on the Position of JQ923422 | |
|---|---|---|---|---|---|
| Sequences | Position on 5′ REH of JQ923422 | ||||
| 5 | 1 | 5283–5451 | GCGCATGTTATGGATTACATCAT | unknown | 196–272 |
| 9 | 14 | 5283–5402 | GCTGATATAAAACT | 5467–5480 | 119–272 |
| 11 | 4 | 5283–5402 | GCTGATATAAAACT | 5467–5480 | 119–272 |
| 13 | 6 | 5283–5426 | ATGTACAACAACAACACATTAAAAGATAT | 5491–5519 | 139–272 |
| 14 | 4 | 5283–5402 | GCTGATATAAAACTAAGATGGCGCATGTAC | 5467–5496 | 112–272 |
| 20 | 1 | 5283–5402 | GCTGATATAAAACTAAGATG | 5467–5486 | 73–272 |
| 21 | 1 | 5283–5402 | GCTGATATAAAACTAAGATG | 5467–5486 | 71–272 |
| 23 | 1 | 5283–5426 | ATGTACAA | 5491–5498 | 72–272 |
| 24 | 2 | 5283–5402 | GCTGATATAAAACTAAGATGGCGCATGTACA | 5467–5497 | 67–272 |
| 28 | 2 | 5283–5442 | AAAGT | unknown | 69–272 |
| 29 | 1 | 5283–5429 | TAGATCA | unknown | 56–272 |
| 31 | 1 | 5283–5444 | GC | 5467–5468 | 48–272 |
| 32 | 1 | 5283–5402 | GCTGATATAAAACTAAGATGGCG | 5467–5489 | 17–272 |
| Variants | Alignment Results between Variants and Reference Sequence | ||
|---|---|---|---|
| 288a | 1–120 5283–5402 | 121–144 5403–5426 | 145–288 129–272 |
| 288b | 1–120 5283–5402 | 121–144 5420–5397 * | 145–288 129–272 |
| 304a | 1–120 5283–5402 | 121–149 5403–5431 | 150–304 118–272 |
| 304b | 1–120 5283–5402 | 121–149 5420–5392 * | 150–304 118–272 |
| 374a | 1–124 5283–5406 | 125–242 23–140 | 243–374 141–272 |
| 374b | 1–124 5283–5406 | 125–242 118–1 * | 243–374 141–272 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; De, R.; Xu, Y.; Han, Z.; Zhu, R.; Sun, Y.; Jia, L.; Chen, D.; Zhou, Y.; Guo, Q.; et al. Diverse Head-to-Tail Sequences in the Circular Genome of Human Bocavirus Genotype 1 among Children with Acute Respiratory Infections Implied the Switch of Template Chain in the Rolling-Circle Replication Model. Pathogens 2024, 13, 757. https://doi.org/10.3390/pathogens13090757
Zhang K, De R, Xu Y, Han Z, Zhu R, Sun Y, Jia L, Chen D, Zhou Y, Guo Q, et al. Diverse Head-to-Tail Sequences in the Circular Genome of Human Bocavirus Genotype 1 among Children with Acute Respiratory Infections Implied the Switch of Template Chain in the Rolling-Circle Replication Model. Pathogens. 2024; 13(9):757. https://doi.org/10.3390/pathogens13090757
Chicago/Turabian StyleZhang, Kexiang, Ri De, Yanpeng Xu, Zhenzhi Han, Runan Zhu, Yu Sun, Liping Jia, Dongmei Chen, Yutong Zhou, Qi Guo, and et al. 2024. "Diverse Head-to-Tail Sequences in the Circular Genome of Human Bocavirus Genotype 1 among Children with Acute Respiratory Infections Implied the Switch of Template Chain in the Rolling-Circle Replication Model" Pathogens 13, no. 9: 757. https://doi.org/10.3390/pathogens13090757