Platelet Aggregation Alterations in Patients with Severe Viral Infection Treated at the Intensive Care Unit: Implications for Mortality Risk




Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Data Collection
2.3. Platelet Aggregation Measurements
2.4. Patient Management
2.5. Statistical Analysis
3. Results
3.1. Characterization of the Patient Cohort
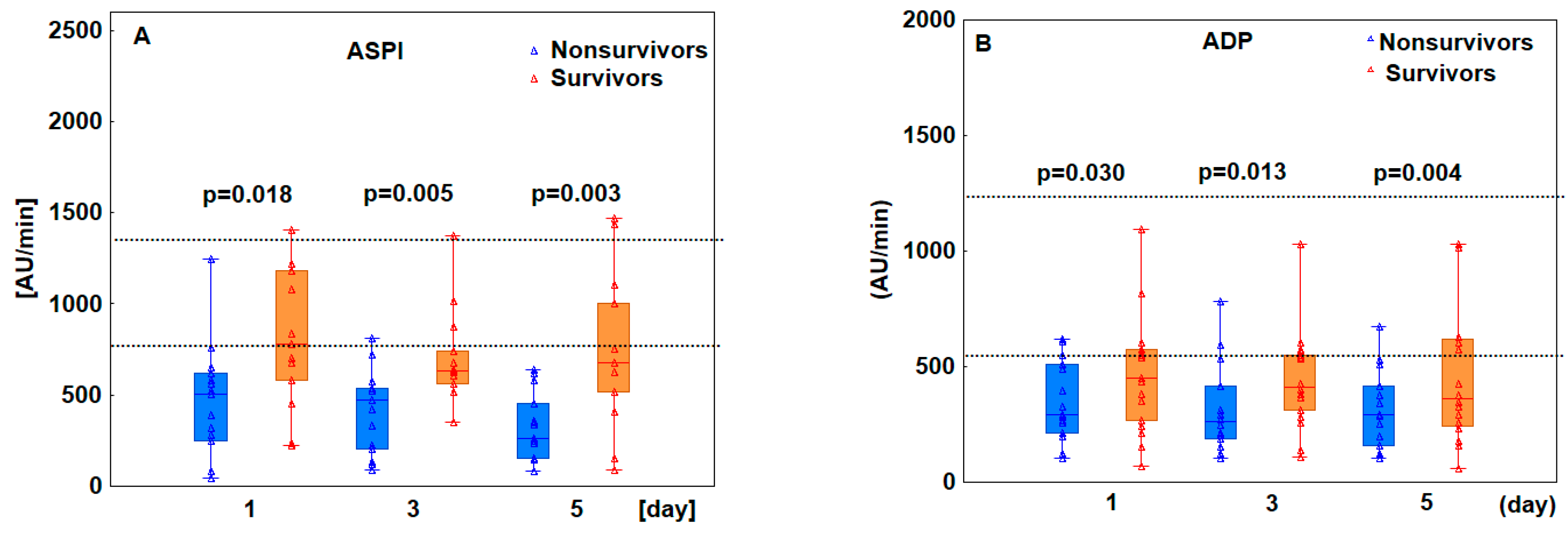
3.2. Patients Who Ultimately Died Exhibited Decreased Platelet Aggregation
3.3. Other Parameters of Coagulation Were Normal except for D-Dimer and Fibrinogen Levels
3.4. Decreased Platelet Aggregation Results Predicted Poor Prognosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Stoppelaar, S.F.; van’t Veer, C.; van der Poll, T. The role of platelets in sepsis. Thromb. Haemost. 2014, 112, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Evtugina, N.G.; Peshkova, A.D.; Safiullina, S.I.; Andrianova, I.A.; Khabirova, A.I.; Nagaswami, C.; Khismatullin, R.R.; Sannikova, S.S.; Weisel, J.W. Altered platelet and coagulation function in moderate-to-severe COVID-19. Sci. Rep. 2021, 11, 16290. [Google Scholar] [CrossRef] [PubMed]
- Sciaudone, A.; Corkrey, H.; Humphries, F.; Koupenova, M. Platelets and SARS-CoV-2 During COVID-19: Immunity, Thrombosis, and Beyond. Circ. Res. 2023, 132, 1272–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Sterne, J.A.C.; Knight, R.; Walker, V.; Ip, S.; Cooper, J.A.; Bolton, T.; Keene, S.; Denholm, R.; Akbari, A.; Abbasizanjani, H.; et al. Circulation Association of COVID-19 With Major Arterial and Venous Thrombotic Diseases: A Population-Wide Cohort Study of 48 Million Adults in England and Wales. Circulation 2022, 146, 892–906. [Google Scholar] [CrossRef]
- Kanth Manne, B.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Ruberto, F.; Chistolini, A.; Curreli, M.; Frati, G.; Marullo, A.G.M.; Biondi-Zoccai, G.; Mancone, M.; Sciarretta, S.; Miraldi, F.; Alessandri, F.; et al. Von Willebrand factor with increased binding capacity is associated with reduced platelet aggregation but enhanced agglutination in COVID-19 patients: Another COVID-19 paradox? J. Thromb. Thrombolysis 2021, 52, 105–110. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 2021, 12, 50. [Google Scholar] [CrossRef]
- Bertini, P.; Guarracino, F.; Falcone, M.; Nardelli, P.; Landoni, G.; Nocci, M.; Paternoster, G. ECMO in COVID-19 Patients: A Systematic Review and Meta-analysis. J. Cardiothorac. Vasc. Anesth. 2022, 36, 2700–2706. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Y.; Liu, J.; Zhou, Z. Thrombosis and bleeding in patients with COVID-19 requiring extracorporeal membrane oxygenation: A systematic review and meta-analysis. Res. Pract. Thromb. Haemost. 2023, 7, 100103. [Google Scholar] [CrossRef]
- Reininger, A.J.; Heijnen, H.F.G.; Schumann, H.; Specht, H.M.; Schramm, W.; Ruggeri, Z.M. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood 2006, 107, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.; Nesbitt, W.S.; Yuan, Y.; Jackson, S.P. Importance of temporal flow gradients and integrin alphaIIbbeta3 mechanotransduction for shear activation of platelets. J. Biol. Chem. 2005, 280, 15430–15437. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Tamura, N.; Li, M.; Handa, M.; Ikeda, Y.; Handa, S.; Ruggri, Z.M. Different effects of various anti-GPIIb-IIIa agents on shear-induced platelet activation and expression of procoagulant activity. J. Thromb. Haemost. 2003, 1, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mondal, N.K.; Zheng, S.; Koenig, S.C.; Slaughter, M.S.; Griffith, B.P.; Wu, Z.J. High Shear Induces Platelet Dysfunction Leading to Enhanced Thrombotic Propensity and Diminished Hemostatic Capacity. Platelets 2019, 30, 112. [Google Scholar] [CrossRef]
- Chen, Z.; Mondal, N.K.; Ding, J.; Gao, J.; Griffith, B.P.; Wu, Z.J. Shear-induced platelet receptor shedding by non-physiological high shear stress with short exposure time: Glycoprotein Ibα and glycoprotein VI. Thromb. Res. 2015, 135, 692. [Google Scholar] [CrossRef]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef]
- Shekar, K.; Badulak, J.; Peek, G.; Boeken, U.; Dalton, H.J.; Arora, L.; Zakhary, B.; Ramanathan, K.; Starr, J.; Akkanti, B.; et al. Extracorporeal Life Support Organization Coronavirus Disease 2019 Interim Guidelines: A Consensus Document from an International Group of Interdisciplinary Extracorporeal Membrane Oxygenation Providers. ASAIO J. 2020, 66, 707–721. [Google Scholar] [CrossRef]
- Papazian, L.; Aubron, C.; Brochard, L.; Chiche, J.D.; Combes, A.; Dreyfuss, D.; Forel, J.M.; Guérin, C.; Jaber, S.; Mekontso-Dessap, A.; et al. Formal guidelines: Management of acute respiratory distress syndrome. Ann. Intensive Care 2019, 9, 69. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Rolfes, V.; Ribeiro, L.S.; Hawwari, I.; Böttcher, L.; Rosero, N.; Maasewerd, S.; Santos, M.L.S.; Próchnicki, T.; Silva, C.M.d.S.; Wanderley, C.W.d.S.; et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep. 2020, 31, 107615. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The inflammatory role of platelets via their TLRs and Siglec receptors. Front. Immunol. 2015, 6, 129756. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Bilaloglu, S.; Cornwell, M.; Burgess, H.M.; Virginio, V.W.; Drenkova, K.; Ibrahim, H.; Yuriditsky, E.; Aphinyanaphongs, Y.; Lifshitz, M.; et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 2021, 19, 3139–3153. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yuan, H.; Wu, Y.L.; Fu, S.; Pan, X.Y. The Predictive Value of Heparin-Binding Protein and D-Dimer in Patients with Sepsis. Int. J. Gen. Med. 2023, 16, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Pan, Y.; Xu, C.; Li, D. Characteristics of emergency patients with markedly elevated D-dimer levels. Sci. Rep. 2020, 10, 7784. [Google Scholar] [CrossRef]
- Rieder, M.; Baldus, N.; Stallmann, D.; Jeserich, M.; Goller, I.; Wirth, L.; Pollmeier, L.; Hofmann, M.; Bode, C.; Busch, H.J.; et al. Early SARS-CoV-2 infection: Platelet-neutrophil complexes and platelet function. Res. Pract. Thromb. Haemost. 2023, 7, 100025. [Google Scholar] [CrossRef]
- Bertolin, A.J.; Dalçóquio, T.F.; Salsoso, R.; Remo, R.H.; Kalil-Filho, R.; Hajjar, L.A.; Siciliano, R.F.; Kallás, E.G.; Baracioli, L.M.; Lima, F.G.; et al. Platelet Reactivity and Coagulation Markers in Patients with COVID-19. Adv. Ther. 2021, 38, 3911–3923. [Google Scholar] [CrossRef]
- Heinz, C.; Miesbach, W.; Herrmann, E.; Sonntagbauer, M.; Raimann, F.J.; Zacharowski, K.; Weber, C.F.; Adam, E.H. Greater Fibrinolysis Resistance but No Greater Platelet Aggregation in Critically Ill COVID-19 Patients. Anesthesiology 2021, 134, 457–467. [Google Scholar] [CrossRef]
- Lipińska-Gediga, M.; Lemańska-Perek, A.; Gozdzik, W.; Adamik, B. Changes in plasma endocan level are related to circulatory but not respiratory failure in critically ill patients with COVID-19. Sci. Rep. 2023, 13, 22307. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.R.; Mills, G.M.; Lawrence, M.; Battle, C.; Morris, K.; Hawkins, K.; Williams, P.R.; Davidson, S.; Thomas, D.; Evans, P.A. The role of whole blood impedance aggregometry and its utilisation in the diagnosis and prognosis of patients with systemic inflammatory response syndrome and sepsis in acute critical illness. PLoS ONE 2014, 9, e108589. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.A.; Larsen, J.B.; Larsen, K.M.; Hvas, A.M. Platelet function in patients with septic shock. Thromb. Res. 2020, 185, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Garcia, C.; Duong, J.A.; Po, M.; Ribes, A.; Payre, B.; Mémier, V.; Sié, P.; Minville, V.; Voisin, S.; Payrastre, B.; et al. Platelet activation and partial desensitization are associated with viral xenophagy in patients with severe COVID-19. Blood Adv. 2022, 6, 3884–3898. [Google Scholar] [CrossRef]
- Maquet, J.; Lafaurie, M.; Sommet, A.; Moulis, G. Thrombocytopenia is independently associated with poor outcome in patients hospitalized for COVID-19. Br. J. Haematol. 2020, 190, e276–e279. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Clark, C.C.; Jukema, B.N.; Barendrecht, A.D.; Spanjaard, J.S.; Jorritsma, N.K.N.; Smits, S.; de Maat, S.; Seinen, C.W.; Verhoef, S.; Parr, N.M.J.; et al. Thrombotic Events in COVID-19 Are Associated with a Lower Use of Prophylactic Anticoagulation Before Hospitalization and Followed by Decreases in Platelet Reactivity. Front. Med. 2021, 8, 650129. [Google Scholar] [CrossRef]
- Fälker, K.; Haglund, L.; Gunnarsson, P.; Nylander, M.; Lindahl, T.L.; Grenegård, M. Protease-activated receptor 1 (PAR1) signalling desensitization is counteracted via PAR4 signalling in human platelets. Biochem. J. 2011, 436, 469–480. [Google Scholar] [CrossRef]
- Tauber, H.; Streif, W.; Fritz, J.; Ott, H.; Weigel, G.; Loacker, L.; Heinz, A.; Velik-Salchner, C. Predicting Transfusion Requirements During Extracorporeal Membrane Oxygenation. J. Cardiothorac. Vasc. Anesth. 2016, 30, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Hoechter, D.J.; Buscher, H.; Venkatesh, K.; Whittam, S.; Joseph, J.; Jansz, P. Prospective observational study of hemostatic alterations during adult extracorporeal membrane oxygenation (ECMO) using point-of-care thromboelastometry and platelet aggregometry. J. Cardiothorac. Vasc. Anesth. 2015, 29, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Kalbhenn, J.; Glonnegger, H.; Büchsel, M.; Priebe, H.J.; Zieger, B. Acquired von Willebrand Syndrome and Desmopressin Resistance During Venovenous Extracorporeal Membrane Oxygenation in Patients With COVID-19: A Prospective Observational Study. Crit. Care Med. 2022, 50, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Balle, C.M.; Jeppesen, A.N.; Christensen, S.; Hvas, A.M. Platelet Function During Extracorporeal Membrane Oxygenation in Adult Patients: A Systematic Review. Front. Cardiovasc. Med. 2018, 5, 157. [Google Scholar] [CrossRef]
- Balle, C.M.; Jeppesen, A.N.; Christensen, S.; Hvas, A.M. Platelet Function During Extracorporeal Membrane Oxygenation in Adult Patients. Front. Cardiovasc. Med. 2019, 6, 114. [Google Scholar] [CrossRef]
- Heuts, S.; Makhoul, M.; Mansouri, A.N.; Taccone, F.S.; Obeid, A.; Belliato, M.; Broman, L.M.; Malfertheiner, M.; Meani, P.; Raffa, G.M.; et al. Defining and understanding the “extra-corporeal membrane oxygenation gap” in the veno-venous configuration: Timing and causes of death. Artif. Organs 2022, 46, 349–361. [Google Scholar] [CrossRef]
- Zeibi Shirejini, S.; Carberry, J.; McQuilten, Z.K.; Burrell, A.J.C.; Gregory, S.D.; Hagemeyer, C.E. Current and future strategies to monitor and manage coagulation in ECMO patients. Thromb. J. 2023, 21, 11. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | All | Survivors | Nonsurvivors | p |
|---|---|---|---|---|
| N = 28 | N = 13 | N = 15 | ||
| Age | 44 (35–55) | 37 (33–45) | 53 (39–57) | 0.664 |
| Gender, female/male (N) | 8/25 | 5/8 | 3/12 | 0.255 |
| Coexisting conditions, n (%) | ||||
| Coronary heart disease | 2 (7) | 0 | 2 (13) | 0.277 |
| Hypertension | 3 (28) | 1 (8) | 2 (13) | 0.555 |
| Diabetes mellitus | 2 (7) | 0 | 2 (13) | 0.277 |
| Stroke | 1 (4) | 1 (8) | 0 | 0.464 |
| Asthma | 2 (7) | 0 | 2 (13) | 0.277 |
| Obesity | 6 (25) | 3 (23) | 4 (27) | 0.587 |
| APACHEII score | 14 (10–18) | 13 (10–15) | 16 (12–21) | 0.169 |
| SOFA score | 9 (8–11) | 9 (8–10) | 10 (7–12) | 0.427 |
| Interleukin 6 (pg/mL) | 47.0 | 20.5 | 110.7 | 0.041 |
| (19.6–214.0) | (12.5–84.7) | (45.3–447.3) | ||
| C-reactive protein (mg/dL) | 142.0 | 146.0 | 141.0 | 0.645 |
| (81.0–217.4) | (67.0–216.0) | (83.0–266.0) | ||
| Septic shock | 21 (75) | 11 (85) | 10 (67) | 0.274 |
| RRT | 4 (14) | 0 | 4 (27) | 0.044 |
| ICU LOS (day) | 23 (16–40) | 30 (24–43) | 18 (12–39) | 0.033 |
| Hospital LOS (day) | 26 (19–48) | 35 (25–59) | 19 (12–45) | 0.050 |
| ASPI < 745 AU/min | ADP < 534 AU/min | TRAP < 941 AU/min | RISTO < 896 AU/min | |
|---|---|---|---|---|
| N (%) | N (%) | N (%) | N (%) | |
| 1 | 19 (68) | 17 (61) | 25 (89) | 19 (68) |
| 3 | 23 (82) | 21 (75) | 26 (93) | 19 (68) |
| 5 | 23 (82) | 21 (75) | 25 (89) | 22 (79) |
| Day | Survivors | Nonsurvivors | p | |
|---|---|---|---|---|
| Platelet count (×109/L) | 1 | 267 (228–309) | 261 (190–358) | 0.747 |
| ref. range 145–400 × 109/L | 3 | 231 (179–272) | 190 (147–258) | 0.197 |
| 5 | 256 (167–267) | 177 (105–221) | 0.105 | |
| D-dimer (mg/L) | 1 | 5.3 (1.9–7.0) | 3.9 (1.8–17.5) | 0.998 |
| ref. range 0–0.5 mg/L | 3 | 2.3 (1.7–4.7) | 6.5 (3.1–11.5) | 0.021 |
| 5 | 4.5 (2.3–6.8) | 10.8 (2.0–15.9) | 0.024 | |
| Fibrinogen (g/L) | 1 | 5.5 (4.2–7.8) | 6.1 (3.2–7.8) | 0.871 |
| ref. range 1.8–3.5 g/L | 3 | 4.4 (4.3–6.7) | 5.8 (3.7–7.1) | 0.644 |
| 5 | 5.9 (4.5–6.7) | 5.9 (4.1–6.9) | 0.844 | |
| AT III (%) | 1 | 93 (73–109) | 85 (78–98) | 0.835 |
| 80–120% | 3 | 80 (70–91) | 80 (69–93) | 0.980 |
| 5 | 80 (71–104) | 88 (66–95) | 0.644 |
| Parameter | Day | Survivors | Nonsurvivors | p |
|---|---|---|---|---|
| EXTEM | ||||
| CT (sec) | 1 | 69 (63–75) | 66 (58–91) | 0.857 |
| ref. range 38–79 s. | 3 | 71 (60–74) | 76 (64–90) | 0.336 |
| 5 | 59 (54–81) | 76 (64–81) | 0.219 | |
| CFT (sec) | 1 | 58 (55–63) | 63 (55–77) | 0.180 |
| ref. range 34–159 s. | 3 | 50 (49–61) | 69 (52–75) | 0.081 |
| 5 | 61 (47–66) | 93 (61–134) | 0.066 | |
| MCF (mm) | 1 | 69 (68–72) | 71 (67–74) | 0.959 |
| ref. range (50–72 mm) | 3 | 71 (68–72) | 68 (65–72) | 0.334 |
| 5 | 69 (62–73) | 65 (55–71) | 0.347 | |
| INTEM | ||||
| CT (sec) | 1 | 246 (206–373) | 242 (160–293) | 0.368 |
| ref. range 100–240 s. | 3 | 228 (203–381) | 209 (186–284) | 0.382 |
| 5 | 198 (197–215) | 199 (152–251) | 0.683 | |
| CFT (sec) | 1 | 66 (60–82) | 63 (56–79) | 0.571 |
| ref. range 30–110 s. | 3 | 67 (59–93) | 71 (60–83) | 0.789 |
| 5 | 63 (59–71) | 89 (64–123) | 0.153 | |
| MCF (mm) | 1 | 68 (66–72) | 70 (66–73) | 0.536 |
| ref. range (70–83 mm) | 3 | 69 (68–72) | 66 (64–73) | 0.594 |
| 5 | 68 (62–72) | 65 (54–71) | 0.268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakowski, W.; Smiechowicz, J.; Dragan, B.; Goździk, W.; Adamik, B. Platelet Aggregation Alterations in Patients with Severe Viral Infection Treated at the Intensive Care Unit: Implications for Mortality Risk. Pathogens 2024, 13, 778. https://doi.org/10.3390/pathogens13090778
Bakowski W, Smiechowicz J, Dragan B, Goździk W, Adamik B. Platelet Aggregation Alterations in Patients with Severe Viral Infection Treated at the Intensive Care Unit: Implications for Mortality Risk. Pathogens. 2024; 13(9):778. https://doi.org/10.3390/pathogens13090778
Chicago/Turabian StyleBakowski, Wojciech, Jakub Smiechowicz, Barbara Dragan, Waldemar Goździk, and Barbara Adamik. 2024. "Platelet Aggregation Alterations in Patients with Severe Viral Infection Treated at the Intensive Care Unit: Implications for Mortality Risk" Pathogens 13, no. 9: 778. https://doi.org/10.3390/pathogens13090778