

Comparative Penicillium spp. Transcriptomics: Conserved Pathways and Processes Revealed in Ungerminated Conidia and during Postharvest Apple Fruit Decay
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Cultures, Isolate Propagation, and Apple Fruit Aquisition
2.2. Characterization of Penicillium spp. Virulence in Apple Fruit
2.3. Sample Harvest and RNA Extraction from Penicillium spp. in Apple Fruit
2.4. In Vitro Penicillium spp. Characterization
2.5. Sample Preparation and RNA Extraction from Ungerminated Conidia
2.6. RNA-Sequencing
2.7. Bioinformatic Analyses and Differential Gene Expression
2.8. Gene Ontology and KEGG Mapping
2.9. Statistical Analysis
3. Results
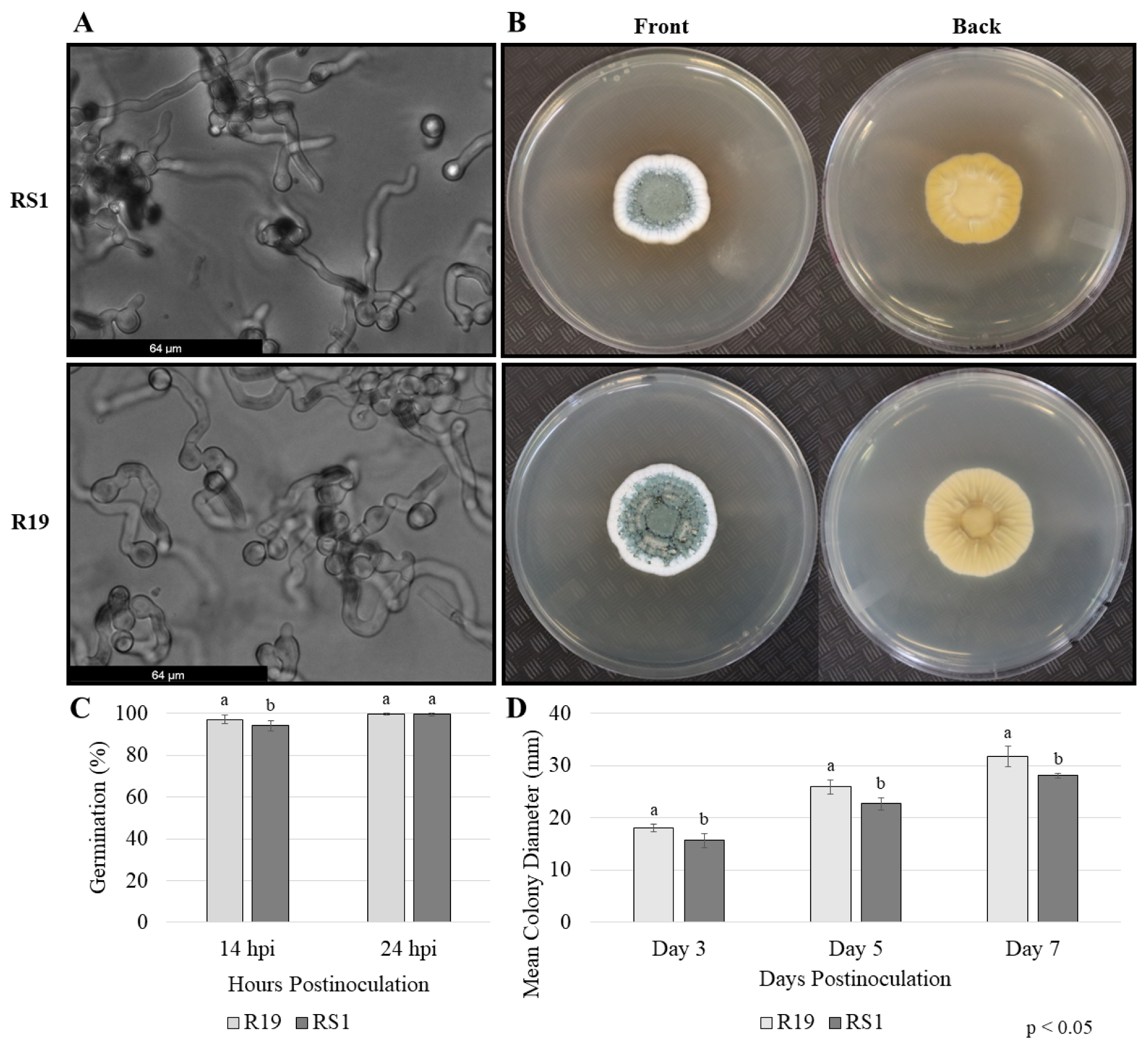
3.1. Disparities in Decay Exist between Two Penicillium spp. in Apple Fruit
3.2. Transcriptomic Differences Were Elucidated between Ungerminated Conidia and Fungal Mycelium during Apple Fruit Decay
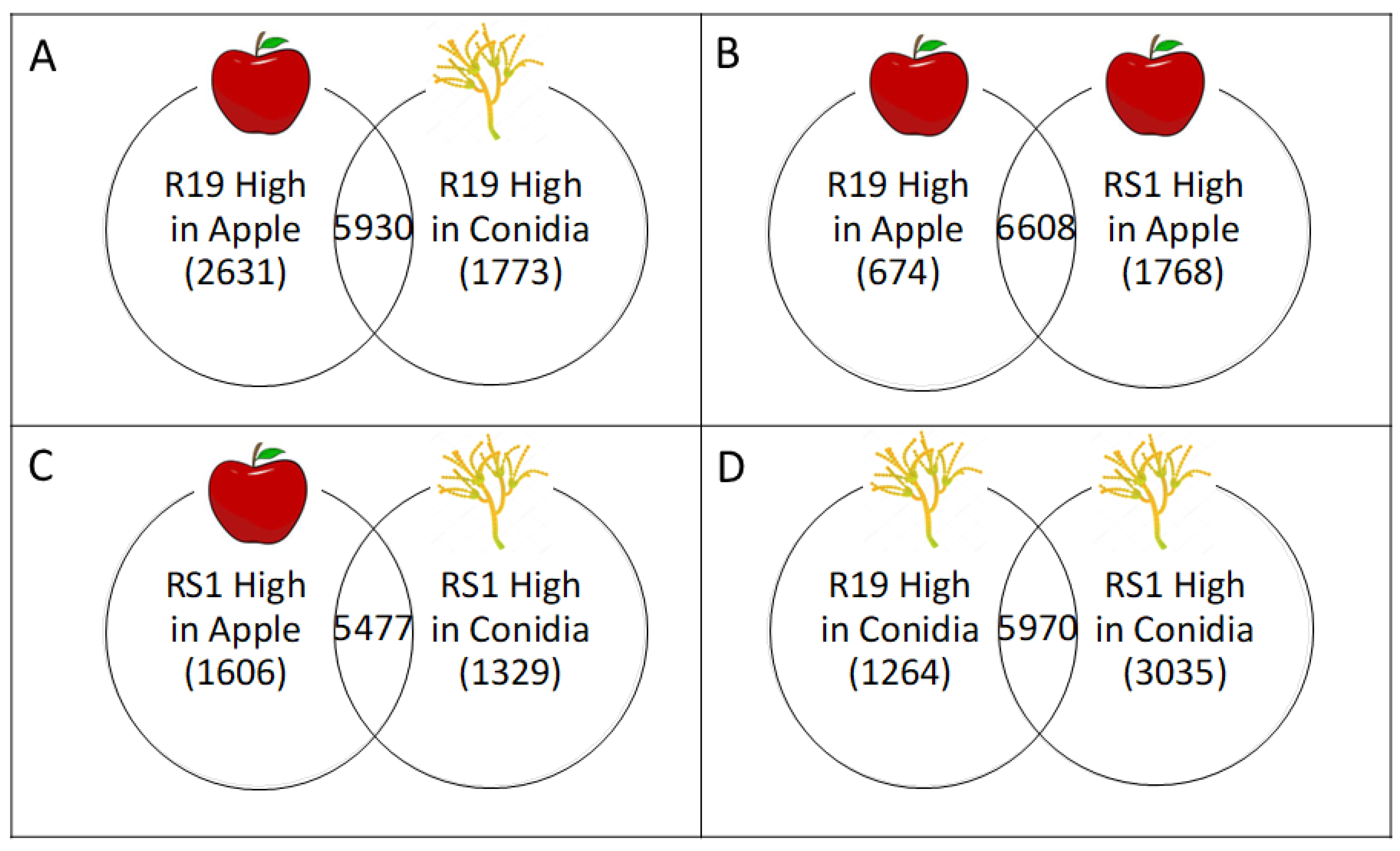
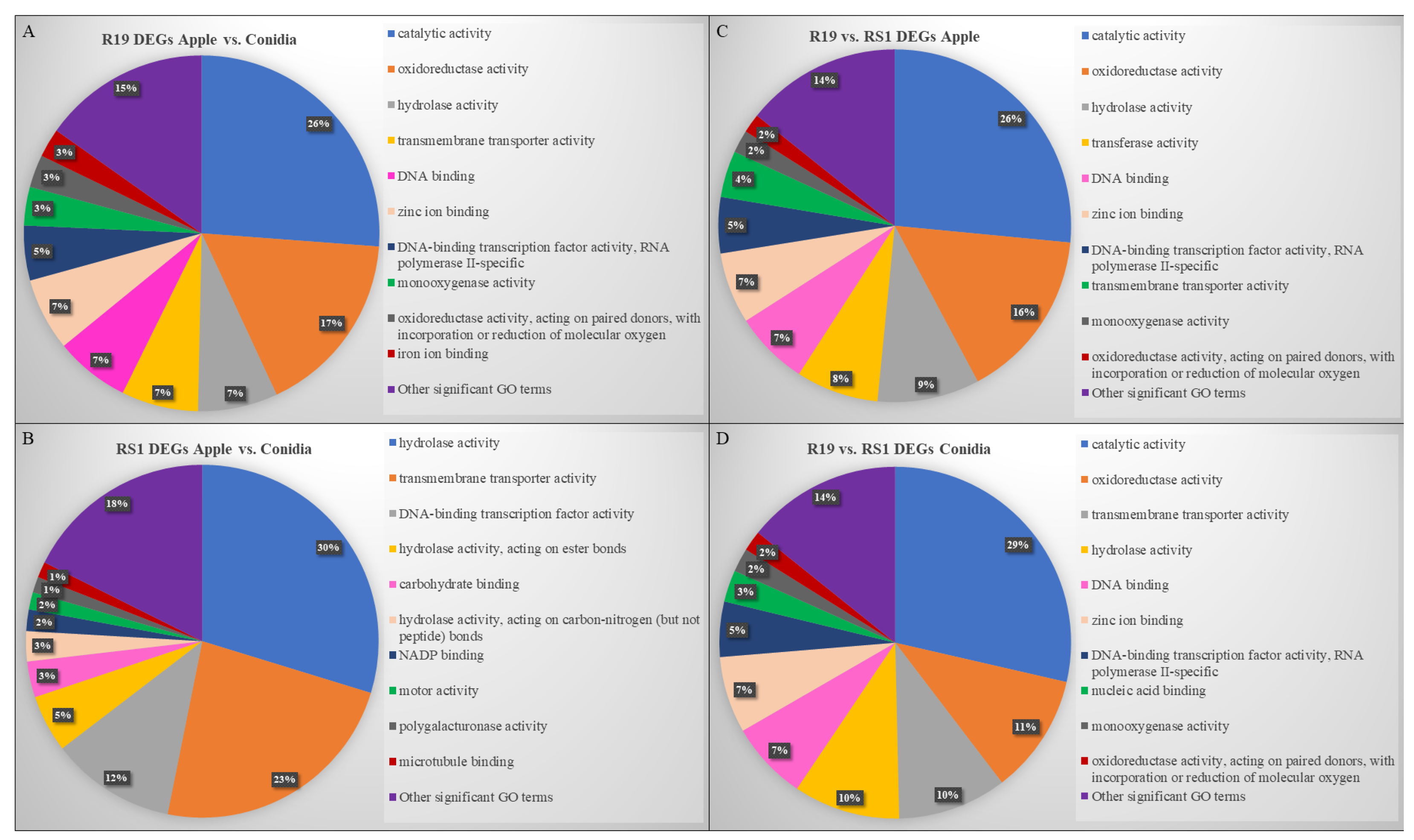
3.3. Overlapping and Distinct Biochemical Themes Are Evident in Different Life Stages Examined in Two Penicillium spp.
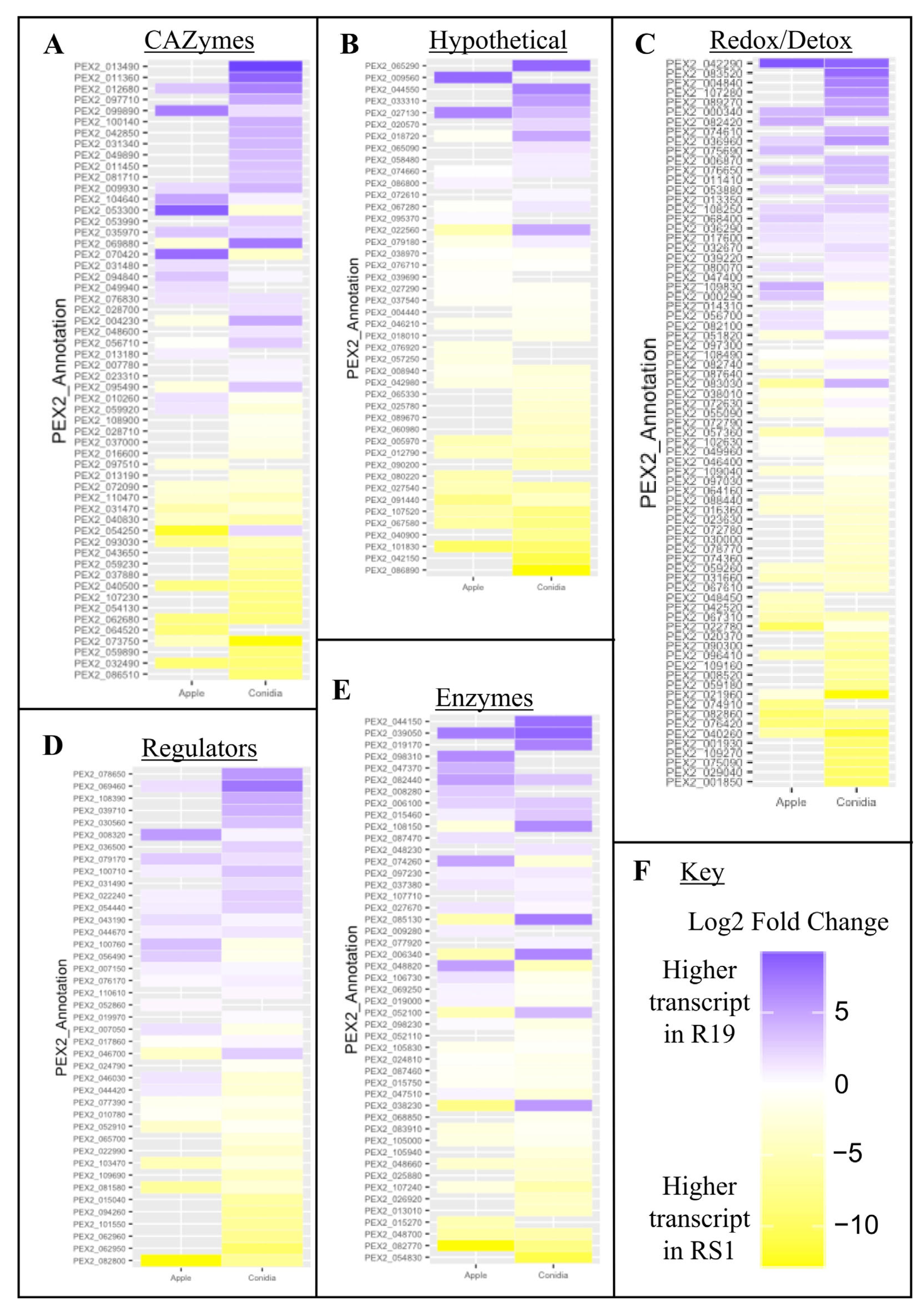
3.4. Fungal Virulence-Associated Genes Encode a Variety of Biochemical Categories
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhong, L.; Carere, J.; Lu, Z.; Lu, F.; Zhou, T. Patulin in Apples and Apple-Based Food Products: The Burdens and the Mitigation Strategies. Toxins 2018, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.I. Penicillium and related genera. In Woodhead Publishing Series in Food Science, Technology and Nutrition, Food Spoilage Microorganisms; de Clive, B.W., Ed.; Woodhead Publishing: Sawston, UK, 2006; pp. 437–450. [Google Scholar] [CrossRef]
- Bradshaw, M.; Bartholomew, H.; Lichtner, F.J.; Gaskins, V.; Jurick, W.M., II. First report of blue mold caused by Penicillium polonicum on apple in the United States. Plant Dis. 2021, 106, 762. [Google Scholar] [CrossRef] [PubMed]
- Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of Penicillium subgenus Penicillium. A guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Stud. Mycol. 2004, 49, 1–174. [Google Scholar]
- Wu, G.; Jurick, W.M., II; Lichtner, F.J.; Peng, H.; Yin, G.; Gaskins, V.L.; Yin, Y.; Hua, S.; Peter, K.A.; Bennett, J.W. Whole-genome Comparisons of Penicillium Spp. Reveals Secondary Metabolic Gene Clusters and Candidate Genes Associated with Fungal Aggressiveness during Apple Fruit Decay. PeerJ 2019, 7, E6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, D.F.; Basham, H.G. Degradation of plant cell walls and membranes by microbial enzymes. Encycl. Plant Physiol. New Ser. 1976, 4, 316–355. [Google Scholar] [CrossRef]
- Bi, F.; Barad, S.; Ment, D.; Luria, N.; Dubey, A.; Casado, V.; Glam, N.; Mínguez, J.D.; Espeso, E.A.; Fluhr, R.; et al. Carbon regulation of environmental pH by secreted small molecules that modulate pathogenicity in phytopathogenic fungi. Mol. Plant Pathol. 2016, 17, 1178–1195. [Google Scholar] [CrossRef] [Green Version]
- Ismaiel, A.A.; Papenbrock, J. Mycotoxins: Producing fungi and mechanisms of phytotoxicity. Agriculture 2015, 5, 492–537. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Conway, W.S.; Sams, C.E. Purification and characterization of a polygalacturonase produced by Penicillium expansum in apple fruit. Phytopathology 1996, 86, 1160–1166. [Google Scholar] [CrossRef]
- Jurick, W.M., II; Vico, I.; Mcevoy, J.; Whitaker, B.; Janisiewicz, W.; Conway, W. Isolation, purification, and characterization of a polygalacturonase produced in Penicillium solitum-decayed ‘Golden Delicious’ apple fruit. Phytopathology 2009, 99, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Li, G.; Liu, J.; Wisniewski, M.; Droby, S.; Levin, E.; Huang, S.; Liu, Y. Multiple transcriptomic analyses and characterization of pathogen-related core effectors and LysM family members reveal their differential roles in fungal growth and pathogenicity in Penicillium expansum. Mol. Genet. Genom. MGG 2020, 295, 1415–1429. [Google Scholar] [CrossRef]
- Levin, E.; Raphael, G.; Ma, J.; Ballester, A.R.; Feygenberg, O.; Norelli, J.; Aly, R.; Gonzalez-Candelas, L.; Wisniewski, M.; Droby, S. Identification and Functional Analysis of NLP-Encoding Genes from the Postharvest Pathogen Penicillium expansum. Microorganisms 2019, 7, 175. [Google Scholar] [CrossRef] [Green Version]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef]
- Selin, C.; De Kievit, T.; Belmonte, M.; Fernando, W. Elucidating the role of effectors in plant-fungal interactions: Progress and challenges. Front. Microbiol. 2016, 7, 600. [Google Scholar] [CrossRef] [Green Version]
- Sorgo, A.G.; Heilmann, C.J.; Brul, S.; de Koster, C.G.; Klis, F.M. Beyond the wall: Candida albicans secret(e)s to survive. FEMS Microbiol. Lett. 2013, 338, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Luciano-Rosario, D.; Keller, N.P.; Jurick, W.M. Penicillium expansum: Biology, omics, and management tools for a global postharvest pathogen causing blue mould of pome fruit. Mol. Plant Pathol. 2020, 21, 1391–1404. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Bartholomew, H.P.; Fonseca, J.M.; Gaskins, V.L.; Prusky, D.; Jurick, W.M., II. Delivering the goods: Fungal secretion modulates virulence during host–pathogen interactions. Fungal Biol. Rev. 2021, 36, 76–86. [Google Scholar] [CrossRef]
- Bartholomew, H.P.; Bradshaw, M.; Jurick, W.M., II; Fonseca, J.M. The Good, the Bad, and the Ugly: Mycotoxin production during postharvest decay and their influence on tritrophic host-pathogen-microbe interactions. Front. Microbiol. 2021, 12, 611881. [Google Scholar] [CrossRef]
- Jurick, W.M., II; Peng, H.; Beard, H.S.; Garrett, W.M.; Lichtner, F.J.; Luciano-Rosario, D.; Macarisin, O.; Liu, Y.; Peter, K.A.; Gaskins, V.L.; et al. Blistering1 Modulates Penicillium expansum Virulence Via Vesicle-mediated Protein Secretion. Mol. Cell. Proteom. MCP 2020, 19, 344–361. [Google Scholar] [CrossRef]
- Zhang, X.; Zong, Y.; Gong, D.; Zhang, F.; Yu, L.; Bi, Y.; Sionov, E.; Prusky, D. Small GTPase RacA is critical for spore growth, patulin accumulation, and virulence of Penicillium expansum. Postharvest Biol. Technol. 2022, 191, 111964. [Google Scholar] [CrossRef]
- Tannous, J.; Barda, O.; Luciano-Rosario, D.; Prusky, D.B.; Sionov, E.; Keller, N.P. New Insight Into Pathogenicity and Secondary Metabolism of the Plant Pathogen Penicillium expansum Through Deletion of the Epigenetic Reader SntB. Front. Microbiol. 2020, 11, 610. [Google Scholar] [CrossRef]
- Kumar, D.; Barad, S.; Chen, Y.; Luo, X.; Tannous, J.; Dubey, A.; Glam Matana, N.; Tian, S.; Li, B.; Keller, N.; et al. LaeA regulation of secondary metabolism modulates virulence in Penicillium expansum and is mediated by sucrose. Mol. Plant Pathol. 2017, 18, 1150–1163. [Google Scholar] [CrossRef]
- Tannous, J.; Kumar, D.; Sela, N.; Sionov, E.; Prusky, D.; Keller, N.P. Fungal attack and host defense pathways unveiled in near-avirulent interactions of Penicillium expansum creA mutants on apples. Mol. Plant Pathol. 2018, 19, 2635–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, B.; Xu, X.; Zhang, Z.; Tian, S. The pH-responsive PacC transcription factor plays pivotal roles in virulence and patulin biosynthesis in Penicillium expansum. Environ. Microbiol. 2018, 20, 4063–4078. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Z.; Li, B.; Tian, S. PeMetR-mediated sulfur assimilation is essential for virulence and patulin biosynthesis in Penicillium expansum. Environ. Microbiol. 2021, 23, 5555–5568. [Google Scholar] [CrossRef] [PubMed]
- El Hajj Assaf, C.; Snini, S.P.; Tadrist, S.; Bailly, S.; Naylies, C.; Oswald, I.P.; Lorber, S.; Puel, O. Impact of veA on the development, aggressiveness, dissemination and secondary metabolism of Penicillium expansum. Mol. Plant Pathol. 2018, 19, 1971–1983. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Chen, Y.; Zong, Y.; Shang, Y.; Zhang, Z.; Xu, X.; Wang, X.; Long, M.; Tian, S. Dissection of patulin biosynthesis, spatial control and regulation mechanism in Penicillium expansum. Environ. Microbiol. 2019, 21, 1124–1139. [Google Scholar] [CrossRef]
- Sánchez-Torres, P.; Vilanova, L.; Ballester, A.R.; López-Pérez, M.; Teixidó, N.; Viñas, I.; Usall, J.; González-Candelas, L.; Torres, R. Unravelling the contribution of the Penicillium expansum PeSte12 transcription factor to virulence during apple fruit infection. Food Microbiol. 2017, 69, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Barad, S.; Horowitz, S.B.; Kobiler, I.; Sherman, A.; Prusky, D. Accumulation of the mycotoxin patulin in the presence of gluconic acid contributes to pathogenicity of Penicillium expansum. Mol. Plant-Microbe Interact. 2013, 27, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zong, Y.; Gong, D.; Yu, L.; Sionov, E.; Bi, Y.; Prusky, D. NADPH Oxidase Regulates the Growth and Pathogenicity of Penicillium expansum. Front. Plant Sci. 2021, 12, 696210. [Google Scholar] [CrossRef]
- Tannous, J.; Snini, S.P.; El Khoury, R.; Canlet, C.; Pinton, P.; Lippi, Y.; Alassane-Kpembi, I.; Gauthier, T.; El Khoury, A.; Atoui, A.; et al. Patulin transformation products and last intermediates in its biosynthetic pathway, E- and Z-ascladiol, are not toxic to human cells. Arch. Toxicol. 2017, 91, 2455–2467. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Li, B.; Tian, S. Arginine Methyltransferase PeRmtC Regulates Development and Pathogenicity of Penicillium expansum via Mediating Key Genes in Conidiation and Secondary Metabolism. J. Fungi 2021, 7, 807. [Google Scholar] [CrossRef]
- Yu, J.; Wu, G.; Jurick, W.M., II; Gaskins, V.L.; Yin, Y.; Yin, G.; Bennett, J.W.; Shelton, D.R. Genome Sequence of Penicillium solitum RS1, Which Causes Postharvest Apple Decay. Genome Announc. 2016, 4, e00363-16. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Jurick, W.M.; Cao, H.; Yin, Y.; Gaskins, V.L.; Losada, L.; Zafar, N.; Kim, M.; Bennett, J.W.; Nierman, W.C. Draft Genome Sequence of Penicillium expansum Strain R19, Which Causes Postharvest Decay of Apple Fruit. Genome Announc. 2014, 2, e00635-14. [Google Scholar] [CrossRef] [Green Version]
- Bartholomew, H.P.; Bradshaw, M.J.; Macarisin, O.; Gaskins, V.L.; Fonseca, J.M.; Jurick, W.M., II. More than a virulence factor: Patulin is a non-host specific toxin that inhibits postharvest phytopathogens and requires efflux for Penicillium tolerance. Phytopathology 2022, 112, 1165–1174. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Simms, D.; Cizdziel, P.; Chomczynski, P. TRIzol: A new reagent for optimal single-step isolation of RNA. Focus 1993, 15, 99–102. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 October 2022).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon: Fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- R Core T. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.r-project.org/ (accessed on 1 October 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. A Publ. Protein Soc. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Xu, M.; Yang, Q.; Adwoa Serwah Boateng, N.; Ahima, J.; Dou, Y.; Zhang, H. Ultrastructure observation and transcriptome analysis of Penicillium expansum invasion in postharvest pears. Postharvest Biol. Technol. 2020, 165, 111198. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, X.; Luo, J.; Ye, B.; Zhou, Y.; Zhou, L.; Lai, T. Identification of differentially expressed genes involved in spore germination of Penicillium expansum by comparative transcriptome and proteome approaches. MicrobiologyOpen 2018, 7, e562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zheng, X.; Zhang, X.; Zhao, L.; Yang, Q.; Boateng, N.; Ahima, J.; Liu, J.; Zhang, H. Comparative Transcriptomic Analysis of the Interaction between Penicillium expansum and Apple Fruit (Malus pumila Mill.) during Early Stages of Infection. Microorganisms 2019, 7, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barad, S.; Sela, N.; Kumar, D.; Kumar-Dubey, A.; Glam-Matana, N.; Sherman, A.; Prusky, D. Fungal and host transcriptome analysis of pH-regulated genes during colonization of apple fruits by Penicillium expansum. BMC Genom. 2016, 17, 330. [Google Scholar] [CrossRef] [Green Version]
- Lichtner, F.J.; Gaskins, V.L.; Cox, K.D.; Jurick, W.M., II. Global transcriptomic responses orchestrate difenoconazole resistance in Penicillium spp. causing blue mold of stored apple fruit. BMC Genom. 2020, 21, 574. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, M.; Leroch, M.; Mosbach, A.; Walker, A.-S.; Fillinger, S.; Mernke, D.; Schoonbeek, H.; Pradier, J.; Leroux, P.; De Waard, M.A.; et al. Fungicide-Driven Evolution and Molecular Basis of Multidrug Resistance in Field Populations of the Grey Mould Fungus Botrytis cinerea. PLoS Pathog. 2009, 5, e1000696. [Google Scholar] [CrossRef]
- Hahn, M. The rising threat of fungicide resistance in plant pathogenic fungi: Botrytis as a case study. J. Chem. Biol. 2014, 7, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Samaras, A.; Ntasiou, P.; Myresiotis, C.; Karaoglanidis, G. Multidrug resistance of Penicillium expansum to fungicides: Whole transcriptome analysis of MDR strains reveals overexpression of efflux transporter genes. Int. J. Food Microbiol. 2020, 335, 108896. [Google Scholar] [CrossRef]
- Yin, G.; Zhao, H.; Pennerman, K.K.; Jurick, W.M., II; Fu, M.; Bu, L.; Guo, A.; Bennett, J.W. Genomic Analyses of Penicillium Species Have Revealed Patulin and Citrinin Gene Clusters and Novel Loci Involved in Oxylipin Production. J. Fungi 2021, 7, 743. [Google Scholar] [CrossRef]
- Sanzani, S.M.; De Girolamo, A.; Schena, L.; Solfrizzo, M.; Ippolito, A.; Visconti, A. Control of Penicillium expansum and patulin accumulation on apples by quercetin and umbelliferone. Eur. Food Res. Technol. 2009, 228, 381–389. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Li, B.; Chen, T.; Tian, S. Reactive oxygen species: A generalist in regulating development and pathogenicity of phytopathogenic fungi. Comput. Struct. Biotechnol. J. 2020, 18, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Diwald, T.T.; Metzler, M. Patulin reduces glutathione level and enzyme activities in rat liver slices. Mol. Nutr. Food Res. 2005, 49, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Bahuguna, A.; Kim, M. The effects of mycotoxin patulin on cells and cellular components. Trends Food Sci. Technol. 2019, 83, 99–113. [Google Scholar] [CrossRef]
- Wang, X.; Jin, C.; Zhong, Y.; Li, X.; Han, J.; Xue, W.; Wu, P.; Xia, X.; Peng, X. Glutathione reduction of patulin-evoked cytotoxicity in HEK293 cells by the prevention of oxidative damage and the mitochondrial apoptotic pathway. J. Agric. Food Chem. 2018, 66, 7775–7785. [Google Scholar] [CrossRef] [PubMed]
- Kuźniak, E.; Urbanek, H. The involvement of hydrogen peroxide in plant responses to stresses. Acta Physiol. Plant 2000, 22, 195–203. [Google Scholar] [CrossRef]
- Barad, S.; Espeso, E.A.; Sherman, A.; Prusky, D. Ammonia modulates P. expansum patulin accumulation. Mol. Plant Pathol. 2016, 17, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Jurick, W.M., II; Vico, I.; Gaskins, V.L.; Garrett, W.M.; Whitaker, B.D.; Janisiewicz, W.J.; Conway, W.S. Purification and biochemical characterization of polygalacturonase produced by Penicillium expansum during postharvest decay of “Anjou” pear. Phytopathology 2010, 100, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewska, M.; Opaliński, Ł.; Veenhuis, M.; van der Klei, I.J. The significance of peroxisomes in secondary metabolite biosynthesis in filamentous fungi. Biotechnol. Lett. 2011, 33, 1921. [Google Scholar] [CrossRef] [Green Version]
- Maggio-Hall, L.A.; Wilson, R.A.; Keller, N.P. Fundamental contribution of beta-oxidation to polyketide mycotoxin production in planta. Mol. Plant-Microbe Interact. 2005, 18, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Peraza-Reyes, L.; Berteaux-Lecellier, V. Peroxisomes and sexual development in fungi. Front. Physiol. 2013, 4, 244. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartholomew, H.P.; Lichtner, F.J.; Bradshaw, M.; Gaskins, V.L.; Fonseca, J.M.; Bennett, J.W.; Jurick, W.M., II. Comparative Penicillium spp. Transcriptomics: Conserved Pathways and Processes Revealed in Ungerminated Conidia and during Postharvest Apple Fruit Decay. Microorganisms 2022, 10, 2414. https://doi.org/10.3390/microorganisms10122414
Bartholomew HP, Lichtner FJ, Bradshaw M, Gaskins VL, Fonseca JM, Bennett JW, Jurick WM II. Comparative Penicillium spp. Transcriptomics: Conserved Pathways and Processes Revealed in Ungerminated Conidia and during Postharvest Apple Fruit Decay. Microorganisms. 2022; 10(12):2414. https://doi.org/10.3390/microorganisms10122414
Chicago/Turabian StyleBartholomew, Holly P., Franz J. Lichtner, Michael Bradshaw, Verneta L. Gaskins, Jorge M. Fonseca, Joan W. Bennett, and Wayne M. Jurick, II. 2022. "Comparative Penicillium spp. Transcriptomics: Conserved Pathways and Processes Revealed in Ungerminated Conidia and during Postharvest Apple Fruit Decay" Microorganisms 10, no. 12: 2414. https://doi.org/10.3390/microorganisms10122414