
A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data

 ,
,
Abstract
:1. Introduction
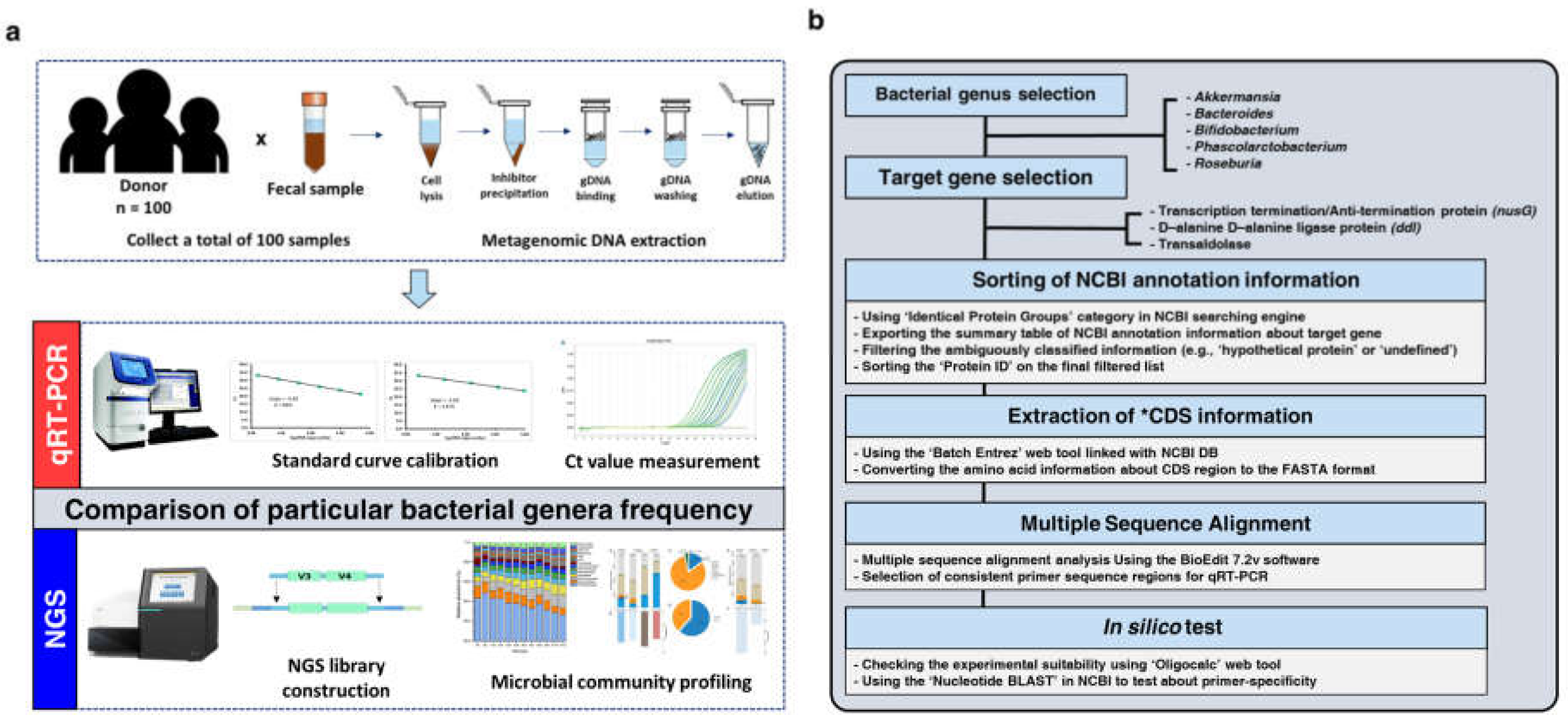
2. Materials and Methods
2.1. Human Stool Samples Collection
2.2. Metagenomic DNA Extraction
2.3. Illumina 16S V3–V4 Amplicon Sequencing Library Preparation and Sequencing
2.4. Bacterial Genus-Specific Primer Design Methods
2.4.1. Bacterial Genera and Target Gene Selection
2.4.2. Sorting of NCBI Annotation Information
2.4.3. Extraction of Coding Sequence Information
2.4.4. Multiple Sequence Alignment and Selection of Target-Specific Regions
2.4.5. In Silico Test
2.5. Bacterial Quantification Using qRT-PCR
2.6. Sanger Sequencing
2.7. 16S V3–V4 Data Processing and Microbial Community Analysis
2.8. Statistical Analysis
3. Results
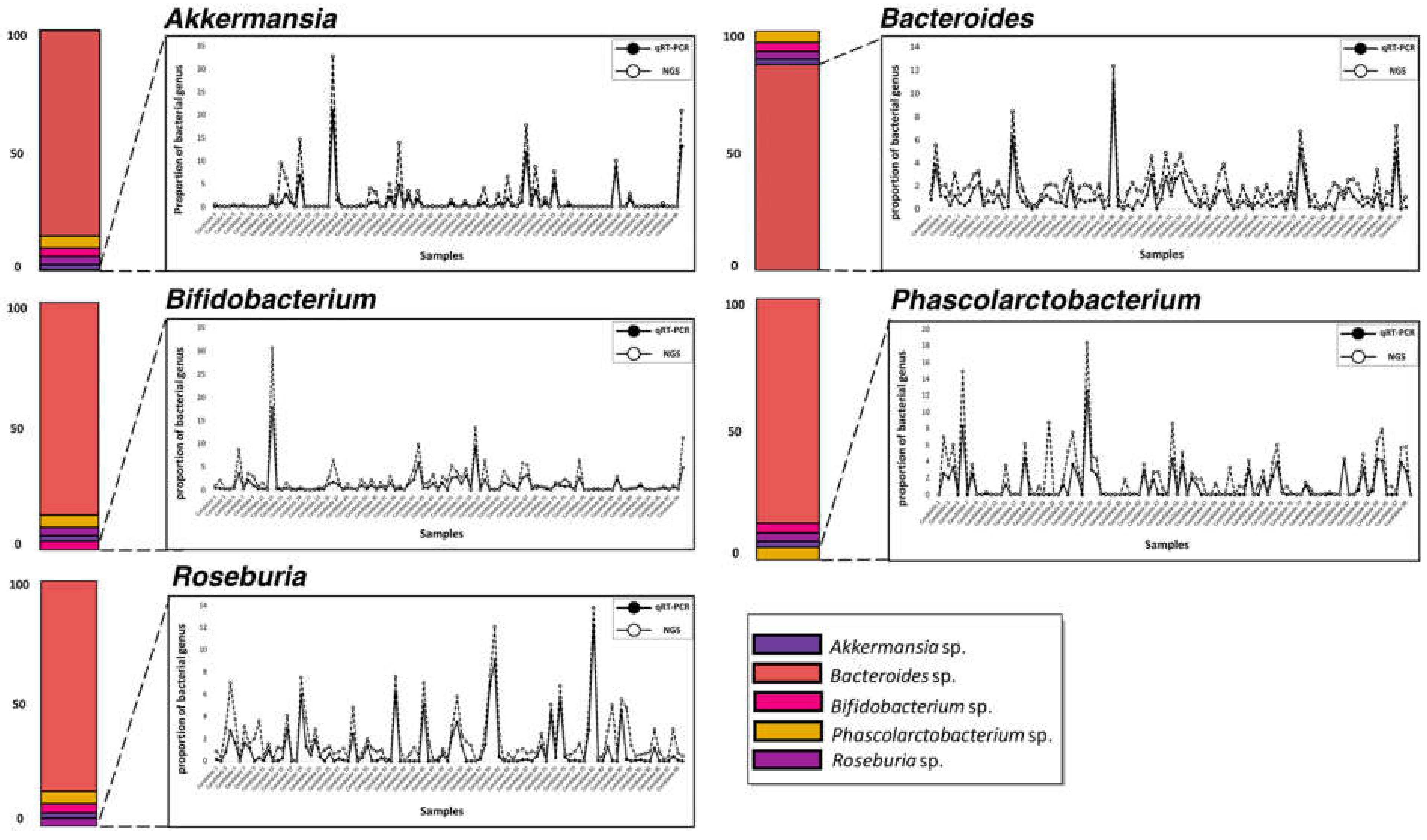
3.1. Selection of Five Bacterial Genera from 16S Metagenome Analysis Data
3.2. Bacterial Genus-Specific Primer Design
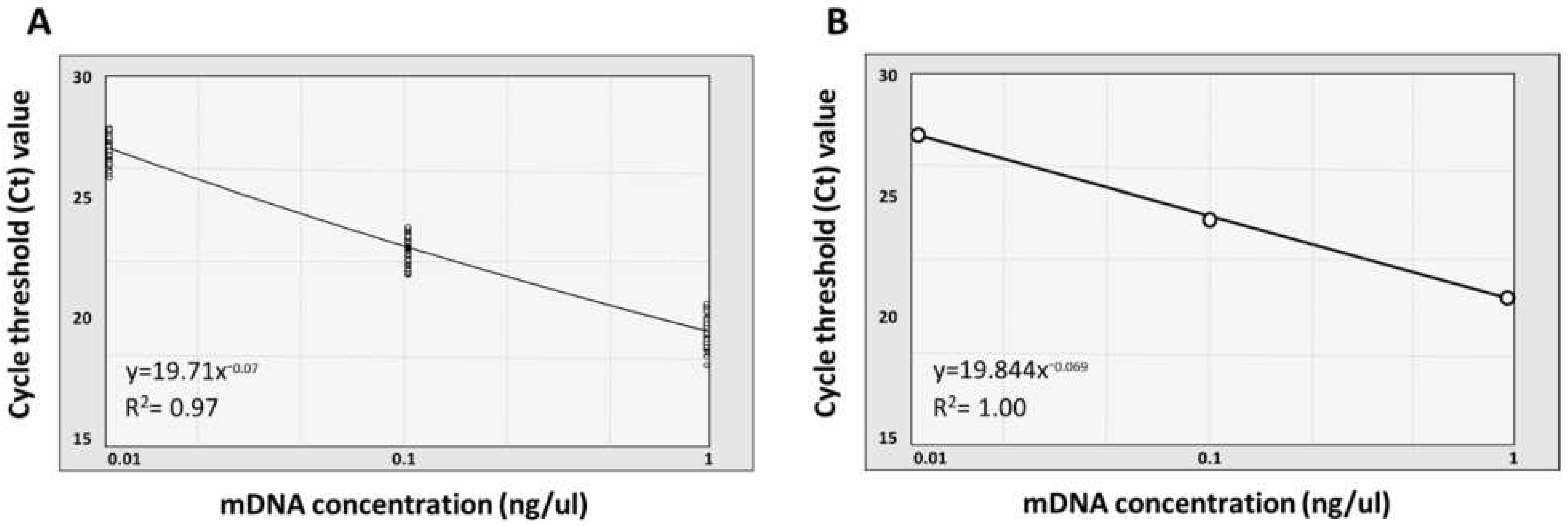
3.3. Quantification and Normalization of Metagenomic DNA
3.4. Parallel Comparison of qRT-PCR and 16S Metagenome Profiling Data
3.5. Verification of Primer Specificity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, K.; Hayashi, M.; Abe, Y.; Kono, M.; Nakajima, K.; Aoyama, Y.; Nishigori, C.; Ishimoto, H.; Ishimatsu, Y.; Nakajima, M.; et al. NGS-based targeted resequencing identified rare subtypes of albinism: Providing accurate molecular diagnosis for Japanese patients with albinism. Pigment Cell Melanoma Res. 2019, 32, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Rim, J.H.; Lee, S.T.; Gee, H.Y.; Lee, B.J.; Choi, J.R.; Park, H.W.; Han, S.H.; Han, J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017, 135, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; Garcia-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Malla, M.A.; Dubey, A.; Kumar, A.; Yadav, S.; Hashem, A.; Abd Allah, E.F. Exploring the Human Microbiome: The Potential Future Role of Next-Generation Sequencing in Disease Diagnosis and Treatment. Front. Immunol. 2018, 9, 2868. [Google Scholar] [CrossRef]
- De Filippis, F.; Parente, E.; Ercolini, D. Recent Past, Present, and Future of the Food Microbiome. Annu. Rev. Food Sci. Technol. 2018, 9, 589–608. [Google Scholar] [CrossRef] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzeby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Feng, G.; Xie, T.; Wang, X.; Bai, J.; Tang, L.; Zhao, H.; Wei, W.; Wang, M.; Zhao, Y. Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 2018, 18, 11. [Google Scholar] [CrossRef]
- Hemmat-Jou, M.H.; Safari-Sinegani, A.A.; Mirzaie-Asl, A.; Tahmourespour, A. Analysis of microbial communities in heavy metals-contaminated soils using the metagenomic approach. Ecotoxicology 2018, 27, 1281–1291. [Google Scholar] [CrossRef]
- Theis, K.R.; Romero, R.; Winters, A.D.; Greenberg, J.M.; Gomez-Lopez, N.; Alhousseini, A.; Bieda, J.; Maymon, E.; Pacora, P.; Fettweis, J.M.; et al. Does the human placenta delivered at term have a microbiota? Results of cultivation, quantitative real-time PCR, 16S rRNA gene sequencing, and metagenomics. Am. J. Obstet. Gynecol. 2019, 220, 267.e1–267.e39. [Google Scholar] [CrossRef] [Green Version]
- Schriefer, A.E.; Cliften, P.F.; Hibberd, M.C.; Sawyer, C.; Brown-Kennerly, V.; Burcea, L.; Klotz, E.; Crosby, S.D.; Gordon, J.I.; Head, R.D. A multi-amplicon 16S rRNA sequencing and analysis method for improved taxonomic profiling of bacterial communities. J. Microbiol. Methods 2018, 154, 6–13. [Google Scholar] [CrossRef]
- Sperling, J.L.; Silva-Brandao, K.L.; Brandao, M.M.; Lloyd, V.K.; Dang, S.; Davis, C.S.; Sperling, F.A.H.; Magor, K.E. Comparison of bacterial 16S rRNA variable regions for microbiome surveys of ticks. Ticks Tick Borne Dis. 2017, 8, 453–461. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lin, H.; Revanna, K.; Dong, Q. A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy. BMC Bioinform. 2017, 18, 247. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Yun, K.; Mun, S.; Chung, W.H.; Choi, S.Y.; Nam, Y.D.; Lim, M.Y.; Hong, C.P.; Park, C.; Ahn, Y.J.; et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci. Rep. 2021, 11, 1727. [Google Scholar] [CrossRef]
- Fujiyoshi, S.; Muto-Fujita, A.; Maruyama, F. Evaluation of PCR conditions for characterizing bacterial communities with full-length 16S rRNA genes using a portable nanopore sequencer. Sci. Rep. 2020, 10, 12580. [Google Scholar] [CrossRef]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sorensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods 2021, 18, 165–169. [Google Scholar] [CrossRef]
- Metras, B.N.; Holle, M.J.; Parker, V.J.; Miller, M.J.; Swanson, K.S. Assessment of commercial companion animal kefir products for label accuracy of microbial composition and quantity. J. Anim. Sci. 2020, 98, skaa301. [Google Scholar] [CrossRef]
- Abrahamson, M.; Hooker, E.; Ajami, N.J.; Petrosino, J.F.; Orwoll, E.S. Successful collection of stool samples for microbiome analyses from a large community-based population of elderly men. Contemp. Clin. Trials Commun. 2017, 7, 158–162. [Google Scholar] [CrossRef]
- Liang, Y.; Dong, T.; Chen, M.; He, L.; Wang, T.; Liu, X.; Chang, H.; Mao, J.H.; Hang, B.; Snijders, A.M.; et al. Systematic Analysis of Impact of Sampling Regions and Storage Methods on Fecal Gut Microbiome and Metabolome Profiles. mSphere 2020, 5, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.M.; Leary, S.D.; Ajami, N.J.; Chipper Keating, S.; Petrosin, J.F.; Hamilton-Shield, J.P.; Gillespie, K.M. Gut microbiome analysis by post: Evaluation of the optimal method to collect stool samples from infants within a national cohort study. PLoS ONE 2019, 14, e0216557. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methe, B.A.; Huttenhower, C. The Human Microbiome Project: A community resource for the healthy human microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef] [Green Version]
- Heiman, M.L.; Greenway, F.L. A healthy gastrointestinal microbiome is dependent on dietary diversity. Mol. Metab. 2016, 5, 317–320. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic Signaling Along The Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2019, 20, 1482. [Google Scholar] [CrossRef] [Green Version]
- DeJong, E.N.; Surette, M.G.; Bowdish, D.M.E. The Gut Microbiota and Unhealthy Aging: Disentangling Cause from Consequence. Cell Host Microbe 2020, 28, 180–189. [Google Scholar] [CrossRef]
- Losso, J.N. Food Processing, Dysbiosis, Gastrointestinal Inflammatory Diseases, and Antiangiogenic Functional Foods or Beverages. Annu. Rev. Food Sci. Technol. 2021, 12, 235–258. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.J.; Giblin, L.; McSweeney, P.L.; Sheehan, J.J.; Cotter, P.D. Nucleic acid-based approaches to investigate microbial-related cheese quality defects. Front. Microbiol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.P.; Peplies, J.; Brettar, I.; Hofle, M.G. Development of a genus-specific next generation sequencing approach for sensitive and quantitative determination of the Legionella microbiome in freshwater systems. BMC Microbiol. 2017, 17, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Mair, R.; Hatcher, C.; Theodore, M.J.; Edmond, K.; Wu, H.M.; Harcourt, B.H.; Carvalho, M.D.G.; Pimenta, F.; Nymadawa, P.; et al. Detection of bacterial pathogens in Mongolia meningitis surveillance with a new real-time PCR assay to detect Haemophilus influenzae. Int. J. Med. Microbiol. 2011, 301, 303–309. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Chen, J.; Shang, S.; Wei, Q.; Yan, J.; Tu, X. Comparison of the fecal microbiota of dholes high-throughput Illumina sequencing of the V3-V4 region of the 16S rRNA gene. Appl. Microbiol. Biotechnol. 2016, 100, 3577–3586. [Google Scholar] [CrossRef]
- Gomez-Gallego, C.; Pohl, S.; Salminen, S.; De Vos, W.M.; Kneifel, W. Akkermansia muciniphila: A novel functional microbe with probiotic properties. Benef. Microbes 2016, 7, 571–584. [Google Scholar] [CrossRef]
- John, G.K.; Mullin, G.E. The Gut Microbiome and Obesity. Curr. Oncol. Rep. 2016, 18, 45. [Google Scholar] [CrossRef]
- Liu, B.; Wang, W.; Zhu, X.; Sun, X.; Xiao, J.; Li, D.; Cui, Y.; Wang, C.; Shi, Y. Response of Gut Microbiota to Dietary Fiber and Metabolic Interaction With SCFAs in Piglets. Front. Microbiol. 2018, 9, 2344. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.A.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Metabolic activities and probiotic potential of bifidobacteria. Int. J. Food Microbiol. 2011, 149, 88–105. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, J.; Zhang, H.; Lee, Y.K.; Zhai, Q.; Chen, W. Roles of intestinal bacteroides in human health and diseases. Crit. Rev. Food Sci. Nutr. 2020, 61, 3518–3536. [Google Scholar] [CrossRef]
- Markowiak-Kopec, P.; Slizewska, K. The Effect of Probiotics on the Production of Short-Chain Fatty Acids by Human Intestinal Microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef]
- Kchouk, M.; Gibrat, J.-F.; Elloumi, M. Generations of Sequencing Technologies: From First to Next Generation. Biol. Med. 2017, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.R.; Ma, W.; Bellecourt, M.J.; Artsimovitch, I.; Martin, A.; Landick, R.; Schulten, K.; Berger, J.M. Mechanism for the Regulated Control of Bacterial Transcription Termination by a Universal Adaptor Protein. Mol. Cell 2018, 71, 911–922.e4. [Google Scholar] [CrossRef] [Green Version]
- Requena, T.; Burton, J.; Matsuki, T.; Munro, K.; Simon, M.A.; Tanaka, R.; Watanabe, K.; Tannock, G.W. Identification, detection, and enumeration of human bifidobacterium species by PCR targeting the transaldolase gene. Appl. Environ. Microbiol. 2002, 68, 2420–2427. [Google Scholar] [CrossRef] [Green Version]
- Ellsworth, B.A.; Tom, N.J.; Bartlett, P.A. Synthesis and evaluation of inhibitors of bacterial d-alanine:d-alanine ligases. Chem. Biol. 1996, 3, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Banihani, S.A. Radish (Raphanus sativus) and Diabetes. Nutrients 2017, 9, 1014. [Google Scholar] [CrossRef] [Green Version]
- Fingas, F.; Volke, D.; Hassert, R.; Fornefett, J.; Funk, S.; Baums, C.G.; Hoffmann, R. Sensitive and immunogen-specific serological detection of Rodentibacter pneumotropicus infections in mice. BMC Microbiol. 2019, 19, 43. [Google Scholar] [CrossRef]
- Pfennig, N.; Bache, R. Selective Isolation of Acetobacterium woodii on Methoxylated Aromatic Acids and Determination of Growth Yields. Arch. Microbiol. 1981, 130, 255–261. [Google Scholar] [CrossRef]
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Tran, Q.; Pham, D.T.; Phan, V. Using 16S rRNA gene as marker to detect unknown bacteria in microbial communities. BMC Bioinform. 2017, 18, 499. [Google Scholar] [CrossRef] [Green Version]
- Zemb, O.; Achard, C.S.; Hamelin, J.; De Almeida, M.L.; Gabinaud, B.; Cauquil, L.; Verschuren, L.M.G.; Godon, J.J. Absolute quantitation of microbes using 16S rRNA gene metabarcoding: A rapid normalization of relative abundances by quantitative PCR targeting a 16S rRNA gene spike-in standard. MicrobiologyOpen 2020, 9, e977. [Google Scholar] [CrossRef]
- Ramsey, P.H. Critical Values for Spearman’s Rank Order Correlation. J. Educ. Stat. 1989, 14, 245–253. [Google Scholar] [CrossRef]
- Freitas, A.C.; Hill, J.E. Bifidobacteria isolated from vaginal and gut microbiomes are indistinguishable by comparative genomics. PLoS ONE 2018, 13, e0196290. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Park, M.S.; Ji, G.E. Improvement of electroporation-mediated transformation efficiency for a Bifidobacterium strain to a reproducibly high level. J. Microbiol. Methods 2019, 159, 112–119. [Google Scholar] [CrossRef]
- Serafini, F.; Turroni, F.; Guglielmetti, S.; Gioiosa, L.; Foroni, E.; Sanghez, V.; Bartolomucci, A.; Motherway, M.O.; Palanza, P.; van Sinderen, D.; et al. An efficient and reproducible method for transformation of genetically recalcitrant bifidobacteria. FEMS Microbiol. Lett. 2012, 333, 146–152. [Google Scholar] [CrossRef] [Green Version]
- The MetaHIT Consortium; Ehrlich, S.D. MetaHIT: The European Union project on metagenomics of the human intestinal tract. In Metagenomics of the Human Body; Springer: Berlin/Heidelberg, Germany, 2010; pp. 307–316. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Rodrigues, C.F.; Stojanovic-Radic, Z.; Dimitrijevic, M.; Aleksic, A.; Neffe-Skocinska, K.; Zielinska, D.; Kolozyn-Krajewska, D.; Salehi, B.; Milton Prabu, S.; et al. Probiotics: Versatile Bioactive Components in Promoting Human Health. Medicina 2020, 56, 433. [Google Scholar] [CrossRef]
- Shukla, P.; Yadav, R. Probiotics for human health: Current progress and applications. In Recent Advances in Applied Microbiology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 133–147. [Google Scholar] [CrossRef]
- Callahan, B.J.; Grinevich, D.; Thakur, S.; Balamotis, M.A.; Yehezkel, T.B. Ultra-accurate microbial amplicon sequencing with synthetic long reads. Microbiome 2021, 9, 130. [Google Scholar] [CrossRef]
- Masco, L.; Vanhoutte, T.; Temmerman, R.; Swings, J.; Huys, G. Evaluation of real-time PCR targeting the 16S rRNA and recA genes for the enumeration of bifidobacteria in probiotic products. Int. J. Food Microbiol. 2007, 113, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Shehata, H.R.; Ragupathy, S.; Shanmughanandhan, D.; Kesanakurti, P.; Ehlinger, T.M.; Newmaster, S.G. Guidelines for Validation of Qualitative Real-Time PCR Methods for Molecular Diagnostic Identification of Probiotics. J. AOAC Int. 2019, 102, 1774–1778. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Taxon | Rank | Target Gene | Foward Primer (5′-3′) | Reverse Primer (5′-3′) | Tm °C (F/R) | GC % (F/R) | Amplicon Size (bp) |
|---|---|---|---|---|---|---|---|
| Akkermansia | genus | ddl | CTTCGTGCTGGAAATCAACACC | CGATAATTCCGCTATTTTTTCGC | 62.1/59.2 | 50/39 | 135 |
| Bacteroides | genus | nusG | GGTGCCTCTCAGACAATCAG | CAATGATACCACTGAATCCGCT | 60.5/60.1 | 55/45 | 149 |
| Bifidobacterium | genus | Transaldolase | AAGGGCATCTCCGTCAACG | GGAGACGAAGAAGGAAGCGA | 59.5/60.5 | 58/55 | 146 |
| Phascolarctobacterium | genus | nusG | TTCCTGGTTATGTGCTTGTAGAG | CAGTCAAAGGAATCGGTTTAGTA | 60.9/59.2 | 43/39 | 114 |
| Roseburia | genus | nusG | AAATACCCGTGGTGTTACCG | GTGTCTCCCTCTGTAAAGTCA | 58.4/59.5 | 50/48 | 130 |
| Dilution Factor | Average * Ct Value | * SD Value | * CV Value | Target Gene |
|---|---|---|---|---|
| 10−3 from 10 ng | 27.31 | 0.49 | 1.78 | 16S rRNA V4 region |
| 10−2 from 10 ng | 23.26 | 0.52 | 2.24 | 16S rRNA V4 region |
| 10−1 from 10 ng | 19.85 | 0.64 | 3.21 | 16S rRNA V4 region |
| Spearman Correlation Test | |||
|---|---|---|---|
| Bacterial Genus | * R Value | Spearman p-Value | * Spearman’s Sig. |
| Akkermansia | 0.895622663 | 2.98 × 10−36 | *** |
| Bacteroides | 0.624122412 | 0 | *** |
| Bifidobacterium | 0.853890597 | 1.51 × 10−29 | *** |
| Phascolarctobacterium | 0.644456804 | 4.67 × 10−13 | *** |
| Roseburia | 0.518642542 | 3.25 × 10−8 | *** |
| Bacterial Genus | Defined Bacterial Taxon Counts in NCBI Database | Defined Bacterial Taxon Rates (%) of Sanger Validation | ||||
|---|---|---|---|---|---|---|
| High Top 5 (Ct Value) | Low Top 5 (Ct Value) | Total | High Top 5 (Ct Value) | Low Top 5 (Ct Value) | Total | |
| Akkermansia | 25 | 25 | 50 | 100.00 | 100.00 | 100.00 |
| Bacteroides | 25 | 24 | 49 | 100.00 | 96.00 | 98.00 |
| Bifidobacterium | 23 | 15 | 38 | 92.00 | 60.00 | 76.00 |
| Phascolarctobacterium | 25 | 25 | 50 | 100.00 | 100.00 | 100.00 |
| Roseburiea | 25 | 25 | 50 | 100.00 | 100.00 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.; Mun, S.; Oh, Y.; Cho, C.-S.; Yun, K.; Ahn, Y.; Chung, W.-H.; Lim, M.Y.; Lee, K.E.; Hwang, T.S.; et al. A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms 2022, 10, 324. https://doi.org/10.3390/microorganisms10020324
Jeong J, Mun S, Oh Y, Cho C-S, Yun K, Ahn Y, Chung W-H, Lim MY, Lee KE, Hwang TS, et al. A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms. 2022; 10(2):324. https://doi.org/10.3390/microorganisms10020324
Chicago/Turabian StyleJeong, Jinuk, Seyoung Mun, Yunseok Oh, Chun-Sung Cho, Kyeongeui Yun, Yongju Ahn, Won-Hyong Chung, Mi Young Lim, Kyung Eun Lee, Tae Soon Hwang, and et al. 2022. "A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data" Microorganisms 10, no. 2: 324. https://doi.org/10.3390/microorganisms10020324
APA StyleJeong, J., Mun, S., Oh, Y., Cho, C.-S., Yun, K., Ahn, Y., Chung, W.-H., Lim, M. Y., Lee, K. E., Hwang, T. S., & Han, K. (2022). A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms, 10(2), 324. https://doi.org/10.3390/microorganisms10020324