
Classification of Environmental Strains from Order to Genus Levels Using Lipid and Protein MALDI-ToF Fingerprintings and Chemotaxonomic Network Analysis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
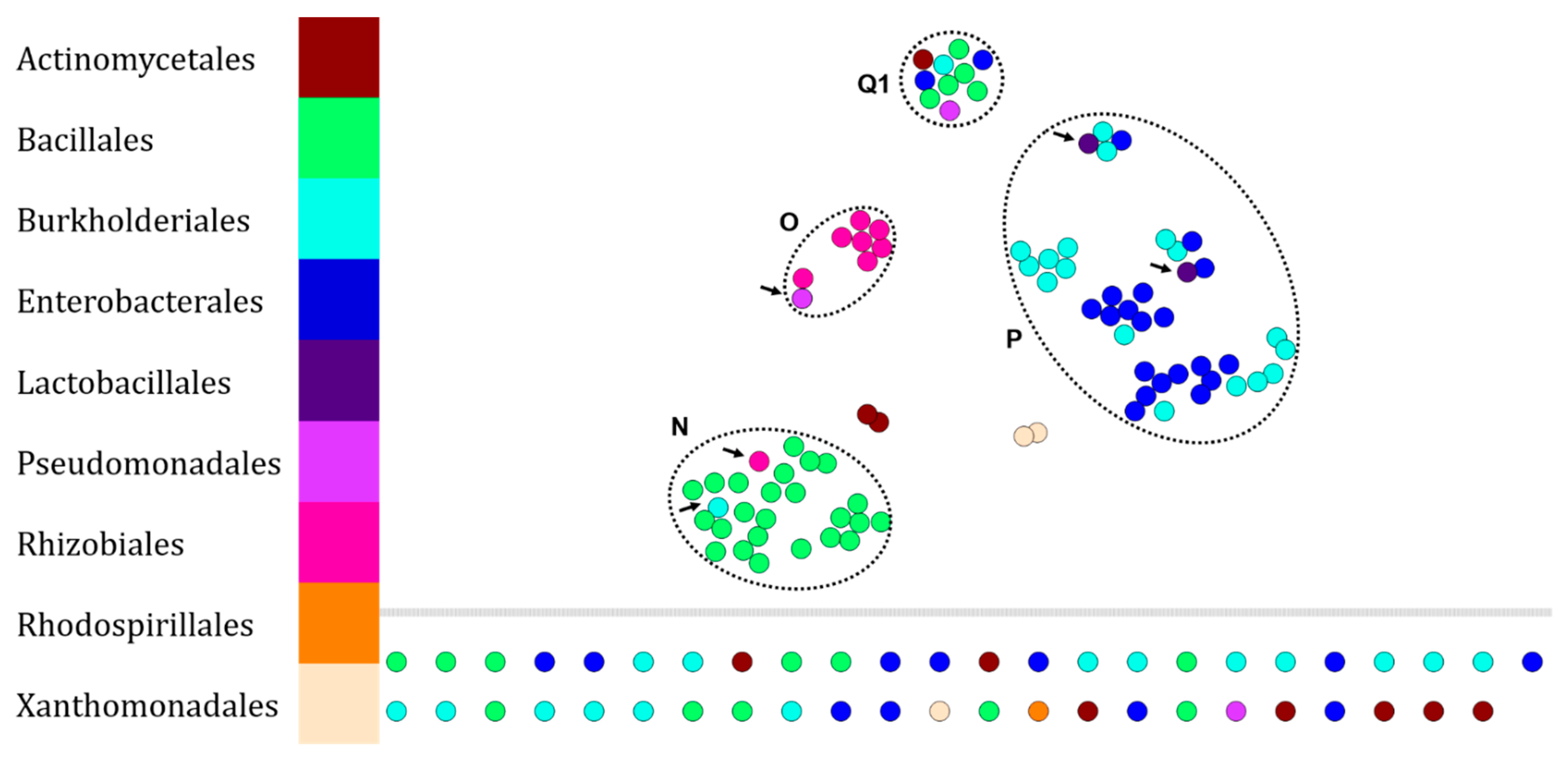
2.1. Proof of Concept: t-SNE Algorithm Clusters Bacterial Protein Fingerprints in a Taxonomy-Consistent Manner
2.2. Robustness of Chemotaxonomic Resolution by Adding Fingerprints of Environmental Isolates to the RKI Dataset
2.3. Differentiation of Environmental Bacteria by MALDI-ToF MS Lipid Fingerprint Analysis
2.4. Differentiation of Environmental Fungi by MALDI-ToF MS Protein Fingerprint Analysis
2.5. Differentiation of Environmental Fungi by MALDI-ToF MS Lipid Fingerprint Analysis
3. Discussion
4. Materials and Methods
4.1. Microorganisms and Cultures
4.2. Identification of Isolates
4.3. Protein and Lipid Extractions
4.4. MALDI-ToF Sample Preparation
4.5. MALDI-ToF Mass Spectrometry
4.6. Spectra Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The Amazing Potential of Fungi: 50 Ways We Can Exploit Fungi Industrially. Fungal Divers 2019, 97, 1–136. [Google Scholar] [CrossRef] [Green Version]
- Katz, L.; Baltz, R.H. Natural Product Discovery: Past, Present, and Future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Briški, F.; Vuković Domanovac, M. Environmental Microbiology. Phys. Sci. Rev. 2017, 2, 20160118. [Google Scholar] [CrossRef]
- Torsvik, V. Prokaryotic Diversity—Magnitude, Dynamics, and Controlling Factors. Science 2002, 296, 1064–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The International Natural Product Sciences Taskforce; Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug. Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Maiden, M.C.J.; van Rensburg, M.J.J.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST Revisited: The Gene-by-Gene Approach to Bacterial Genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Sawana, A.; Adeolu, M.; Gupta, R.S. Molecular Signatures and Phylogenomic Analysis of the Genus Burkholderia: Proposal for Division of This Genus into the Emended Genus Burkholderia Containing Pathogenic Organisms and a New Genus Paraburkholderia Gen. Nov. Harboring Environmental Species. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Woese, C.R.; Fox, G.E.; Pechman, K.R. Comparative Cataloging of 16S Ribosomal Ribonucleic Acid: Molecular Approach to Procaryotic Systematics. Int. J. Syst. Evol. Microbiol. 1977, 27, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S RRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List; Bolchacova, E.; et al. Nuclear Ribosomal Internal Transcribed Spacer (ITS) Region as a Universal DNA Barcode Marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Croxatto, A.; Prod’hom, G.; Greub, G. Applications of MALDI-TOF Mass Spectrometry in Clinical Diagnostic Microbiology. FEMS Microbiol. Rev. 2012, 36, 380–407. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-S.; Kim, Y.H. Rapid and Robust MALDI-TOF MS Techniques for Microbial Identification: A Brief Overview of Their Diverse Applications. J. Microbiol. 2018, 56, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.C.; Hildenbrand, Z.L.; Schug, K.A. Applications of MALDI-TOF MS in Environmental Microbiology. Analyst 2016, 141, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Kallow, W. Differentiation of Indoor Wood Decay Fungi with MALDI-TOF Mass Spectrometry. Holzforschung 2005, 59, 374–377. [Google Scholar] [CrossRef]
- Clark, A.E.; Kaleta, E.J.; Arora, A.; Wolk, D.M. Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry: A Fundamental Shift in the Routine Practice of Clinical Microbiology. Clin. Microbiol. Rev. 2013, 26, 547–603. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.S.; Clark, C.M.; Ómarsdóttir, S.; Sanchez, L.M.; Murphy, B.T. Minimizing Taxonomic and Natural Product Redundancy in Microbial Libraries Using MALDI-TOF MS and the Bioinformatics Pipeline IDBac. J. Nat. Prod. 2019, 82, 2167–2173. [Google Scholar] [CrossRef]
- Sandrin, T.R.; Goldstein, J.E.; Schumaker, S. MALDI TOF MS Profiling of Bacteria at the Strain Level: A Review. Mass Spectrom. Rev. 2013, 32, 188–217. [Google Scholar] [CrossRef]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-Cell MALDI-TOF MS Versus 16S RRNA Gene Analysis for Identification and Dereplication of Recurrent Bacterial Isolates. Front. Microbiol. 2018, 9, 1294. [Google Scholar] [CrossRef]
- Ghyselinck, J.; Van Hoorde, K.; Hoste, B.; Heylen, K.; De Vos, P. Evaluation of MALDI-TOF MS as a Tool for High-Throughput Dereplication. J. Microbiol. Methods 2011, 86, 327–336. [Google Scholar] [CrossRef]
- Bull, A.T.; Goodfellow, M.; Slater, J.H. Biodiversity as a source of innovation in biotechnology. Annu. Rev. Microbiol. 1992, 46, 219–252. [Google Scholar] [CrossRef]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of Small Molecules Using Accurate Mass MS/MS Search. Mass Spec. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.-L.; Litaudon, M.; Touboul, D.; Queiroz, E.F. Innovative Omics-Based Approaches for Prioritisation and Targeted Isolation of Natural Products—New Strategies for Drug Discovery. Nat. Prod. Rep. 2019, 36, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.-F.; Boutet-Mercey, S.; Cachet, X.; De La Torre, E.; Laboureur, L.; Gallard, J.-F.; Retailleau, P.; Brunelle, A.; Dorrestein, P.C.; Costa, J.; et al. Environmentally Friendly Procedure Based on Supercritical Fluid Chromatography and Tandem Mass Spectrometry Molecular Networking for the Discovery of Potent Antiviral Compounds from Euphorbia Semiperfoliata. J. Nat. Prod. 2017, 80, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass Spectral Molecular Networking of Living Microbial Colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [Green Version]
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the T-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef]
- Elie, N.; Santerre, C.; Touboul, D. Generation of a Molecular Network from Electron Ionization Mass Spectrometry Data by Combining MZmine2 and MetGem Software. Anal. Chem. 2019, 91, 11489–11492. [Google Scholar] [CrossRef] [Green Version]
- Dumolin, C.; Aerts, M.; Verheyde, B.; Schellaert, S.; Vandamme, T.; Van der Jeugt, F.; De Canck, E.; Cnockaert, M.; Wieme, A.D.; Cleenwerck, I.; et al. Introducing SPeDE: High-Throughput Dereplication and Accurate Determination of Microbial Diversity from Matrix-Assisted Laser Desorption–Ionization Time of Flight Mass Spectrometry Data. mSystems 2019, 4, e00437-19. [Google Scholar] [CrossRef] [Green Version]
- Dumolin, C.; Peeters, C.; De Canck, E.; Boon, N.; Vandamme, P. Network Analysis Based on Unique Spectral Features Enables an Efficient Selection of Genomically Diverse Operational Isolation Units. Microorganisms 2021, 9, 416. [Google Scholar] [CrossRef]
- Rahi, P.; Prakash, O.; Shouche, Y.S. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass-Spectrometry (MALDI-TOF MS) Based Microbial Identifications: Challenges and Scopes for Microbial Ecologists. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Cassagne, C.; Normand, A.-C.; L’Ollivier, C.; Ranque, S.; Piarroux, R. Performance of MALDI-TOF MS Platforms for Fungal Identification. Mycoses 2016, 59, 678–690. [Google Scholar] [CrossRef]
- Cassagne, C.; Ranque, S.; Normand, A.-C.; Fourquet, P.; Thiebault, S.; Planard, C.; Hendrickx, M.; Piarroux, R. Mould Routine Identification in the Clinical Laboratory by Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry. PLoS ONE 2011, 6, e28425. [Google Scholar] [CrossRef] [PubMed]
- Stübiger, G.; Wuczkowski, M.; Mancera, L.; Lopandic, K.; Sterflinger, K.; Belgacem, O. Characterization of Yeasts and Filamentous Fungi Using MALDI Lipid Phenotyping. J. Microbiol. Methods 2016, 130, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, M.; Guérineau, V.; Genta-Jouve, G.; Roy, M.; Chave, J.; Guillot, R.; Pellissier, L.; Wolfender, J.-L.; Stien, D.; Eparvier, V.; et al. Identification and Dereplication of Endophytic Colletotrichum Strains by MALDI TOF Mass Spectrometry and Molecular Networking. Sci. Rep. 2020, 10, 19788. [Google Scholar] [CrossRef] [PubMed]
- Brel, O.; Touré, S.; Levasseur, M.; Lechat, C.; Pellissier, L.; Wolfender, J.-L.; Van-Elslande, E.; Litaudon, M.; Dusfour, I.; Stien, D.; et al. Paecilosetin Derivatives as Potent Antimicrobial Agents from Isaria Farinosa. J. Nat. Prod. 2020, 83, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Hebra, T.; Elie, N.; Poyer, S.; Van Elslande, E.; Touboul, D.; Eparvier, V. Dereplication, Annotation, and Characterization of 74 Potential Antimicrobial Metabolites from Penicillium Sclerotiorum Using t-SNE Molecular Networks. Metabolites 2021, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Mai, P.-Y.; Levasseur, M.; Buisson, D.; Touboul, D.; Eparvier, V. Identification of Antimicrobial Compounds from Sandwithia Guyanensis-Associated Endophyte Using Molecular Network Approach. Plants 2019, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.T.; de Bel, A.; Martiny, D.; Ranque, S.; Piarroux, R.; Cassagne, C.; Detandt, M.; Hendrickx, M. Identification of Filamentous Fungi Isolates by MALDI-TOF Mass Spectrometry: Clinical Evaluation of an Extended Reference Spectra Library. Med. Mycol. 2014, 52, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Mancini, V.; Dapporto, L.; Baracchi, D.; Luchi, N.; Turillazzi, S.; Capretti, P. Phenotypic Characterization of Cryptic Diplodia Species by MALDI-TOF MS and the Bias of Mycelium Age. For. Path. 2013, 43, 455–461. [Google Scholar] [CrossRef]
- Calvano, C.D.; Zambonin, C.G.; Palmisano, F. Lipid Fingerprinting of Gram-Positive Lactobacilli by Intact Cells—Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Using a Proton Sponge Based Matrix: Lipid Fingerprinting of Gram-Positive Lactobacilli by Intact Cells. Rapid Commun. Mass Spectrom. 2011, 25, 1757–1764. [Google Scholar] [CrossRef]
- Lasch, P.; Stämmler, M.; Schneider, A. Version 3 (20181130) of the MALDI-TOF Mass Spectrometry Database for Identification and Classification of Highly Pathogenic Microorganisms from the Robert Koch-Institute (RKI) 2018. Zenodo. Available online: https://zenodo.org/record/1880975#.Ylt20DURXIU (accessed on 15 February 2019). [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open Source Software for Rapid Proteomics Tools Development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Gibb, S.; Strimmer, K. MALDIquant: A Versatile R Package for the Analysis of Mass Spectrometry Data. Bioinformatics 2012, 28, 2270–2271. [Google Scholar] [CrossRef] [PubMed]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levasseur, M.; Hebra, T.; Elie, N.; Guérineau, V.; Touboul, D.; Eparvier, V. Classification of Environmental Strains from Order to Genus Levels Using Lipid and Protein MALDI-ToF Fingerprintings and Chemotaxonomic Network Analysis. Microorganisms 2022, 10, 831. https://doi.org/10.3390/microorganisms10040831
Levasseur M, Hebra T, Elie N, Guérineau V, Touboul D, Eparvier V. Classification of Environmental Strains from Order to Genus Levels Using Lipid and Protein MALDI-ToF Fingerprintings and Chemotaxonomic Network Analysis. Microorganisms. 2022; 10(4):831. https://doi.org/10.3390/microorganisms10040831
Chicago/Turabian StyleLevasseur, Marceau, Téo Hebra, Nicolas Elie, Vincent Guérineau, David Touboul, and Véronique Eparvier. 2022. "Classification of Environmental Strains from Order to Genus Levels Using Lipid and Protein MALDI-ToF Fingerprintings and Chemotaxonomic Network Analysis" Microorganisms 10, no. 4: 831. https://doi.org/10.3390/microorganisms10040831