Salmonella Enteritidis GalE Protein Inhibits LPS-Induced NLRP3 Inflammasome Activation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Construction of Transposon Mutant Library and Transposon Insertion Sequencing
2.3. Mice and Cell Culture
2.4. Salmonella Infection
2.5. Adhesion and Invasion Assays
2.6. Cytotoxicity and Cytokine Analysis
2.7. Western Blot Analysis
2.8. Fluorescence Resonance Energy Transfer (FRET)
2.9. RNA Sequencing Analysis
2.10. Quantitative Real-Time PCR Analysis
2.11. In Vivo Virulence Assay
2.12. Statistical Analysis
3. Results
3.1. Screening Candidate Salmonella Genes That Modulate Inflammasome Activation
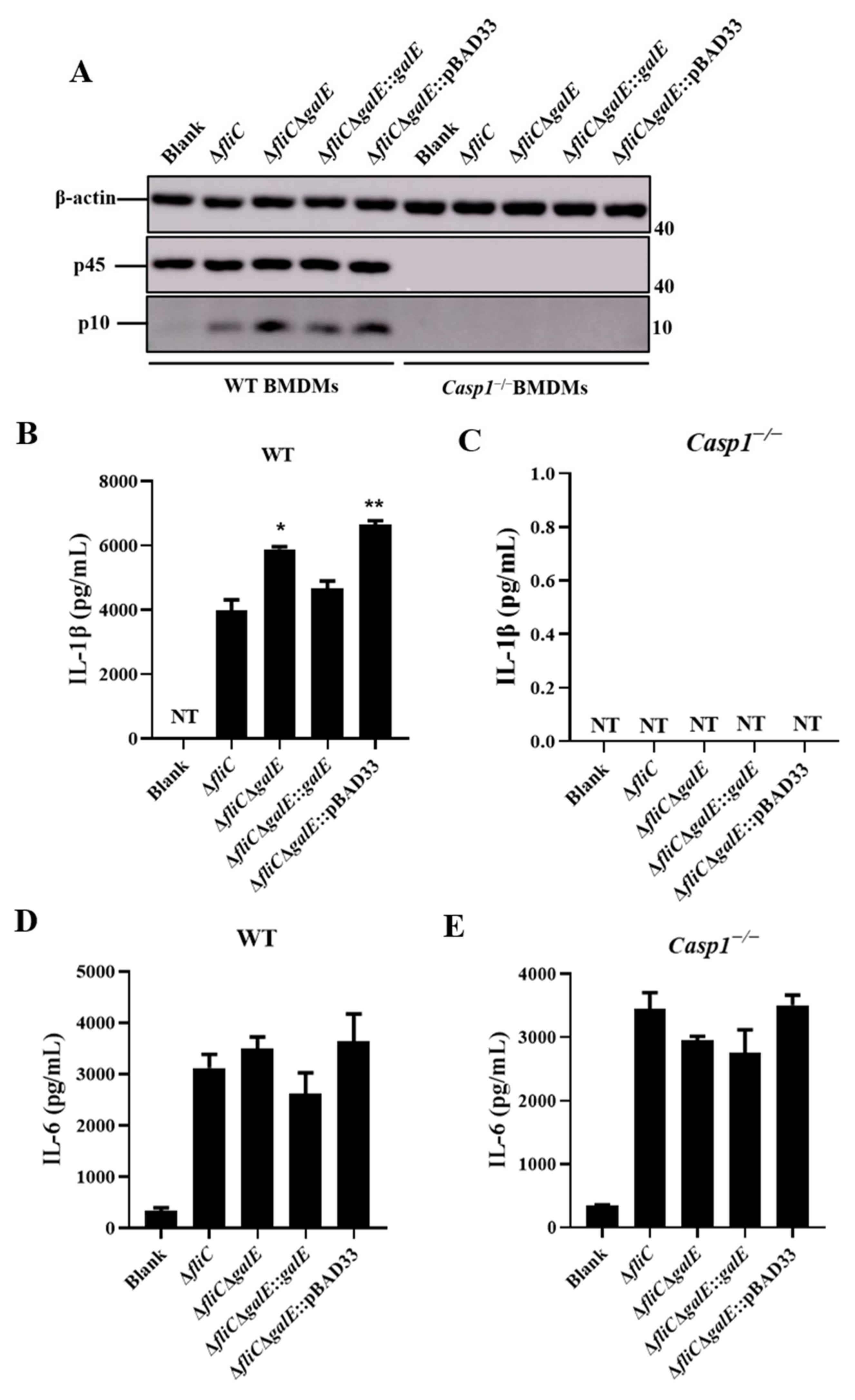
3.2. S. Enteritidis GalE Inhibits the Activation of Inflammasomes
3.3. S. Enteritidis GalE Inhibits the Activation of Caspase-1 in Macrophages via the NLRP3 Inflammasome
3.4. GalE Protein Is Secreted in HeLa Cells
3.5. Identification of Inflammasome Activation Pathways Inhibited by the GalE Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O’Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M.; Int Collaboration Enteric Dis, B. The Global Burden of Nontyphoidal Salmonella Gastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besser, J.M. Salmonella epidemiology: A whirlwind of change. Food Microbiol. 2018, 71, 55–59. [Google Scholar] [CrossRef]
- Brenner, F.W.; Villar, R.G.; Angulo, F.J.; Tauxe, R.; Swaminathan, B. Salmonella nomenclature—Guest commentary. J. Clin. Microbiol. 2000, 38, 2465–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popoff, M.Y.; Bockemuhl, J.; Brenner, F.W. Supplement 1998 (no. 42) to the Kauffmann-White scheme. Res. Microbiol. 2000, 151, 63–65. [Google Scholar] [CrossRef]
- Ferrari, R.G.; Rosario, D.K.A.; Cunha-Neto, A.; Mano, S.B.; Figueiredo, E.E.S.; Conte-Junior, C.A. Worldwide Epidemiology of Salmonella Serovars in Animal-Based Foods: A Meta-analysis. Appl. Environ. Microbiol. 2019, 85, e00591-19. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vargas, F.M.; Abu-El-Haija, M.A.; Gomez-Duarte, O.G. Salmonella infections: An update on epidemiology, management, and prevention. Travel Med. Infect. Dis. 2011, 9, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Eguale, T.; Gebreyes, W.A.; Asrat, D.; Alemayehu, H.; Gunn, J.S.; Engidawork, E. Non-typhoidal Salmonella serotypes, antimicrobial resistance and co-infection with parasites among patients with diarrhea and other gastrointestinal complaints in Addis Ababa, Ethiopia. BMC Infect. Dis. 2015, 15, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraga, A.; Ohlson, M.B.; Miller, S.I. Salmonella e interplay with host cells. Nat. Rev. Microbiol. 2008, 6, 53–66. [Google Scholar] [CrossRef]
- Niedergang, F.; Sirard, J.C.; Blanc, C.T.; Kraehenbuhl, J.P. Entry and survival of Salmonella typhimurium in dendritic cells and presentation of recombinant antigens do not require macrophage-specific virulence factors. Proc. Natl. Acad. Sci. USA 2000, 97, 14650–14655. [Google Scholar] [CrossRef] [Green Version]
- Stapels, D.A.C.; Hill, P.W.S.; Westermann, A.J.; Fisher, R.A.; Thurston, T.L.; Saliba, A.-E.; Blommestein, I.; Vogel, J.; Helaine, S. Salmonella persisters undermine host immune defenses during antibiotic treatment. Science 2018, 362, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Foster, N.; Jones, M.A.; Barrow, P.A. Model of Persistent Salmonella Infection: Salmonella enterica Serovar Pullorum Modulates the Immune Response of the Chicken from a Th17-Type Response towards a Th2-Type Response. Infect. Immun. 2018, 86, e00307-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mon, K.K.Z.; Kern, C.; Chanthavixay, G.; Wang, Y.; Zhou, H. Tolerogenic Immunoregulation towards Salmonella Enteritidis Contributes to Colonization Persistence in Young Chicks. Infect. Immun. 2021, 89, e0073620. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella Infection Induces Recruitment of Caspase-8 to the Inflammasome To Modulate IL-1 beta Production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Naseer, N.; Egan, M.S.; Reyes Ruiz, V.M.; Scott, W.P.; Hunter, E.N.; Demissie, T.; Rauch, I.; Brodsky, I.E.; Shin, S. Human NAIP/NLRC4 and NLRP3 inflammasomes detect Salmonella type III secretion system activities to restrict intracellular bacterial replication. PLoS Pathog. 2022, 18, e1009718. [Google Scholar] [CrossRef]
- Guo, Y.; Gu, D.; Huang, T.; Cao, L.; Zhu, X.; Zhou, Y.; Wang, K.; Kang, X.; Meng, C.; Jiao, X.; et al. Essential role of Salmonella Enteritidis DNA adenine methylase in modulating inflammasome activation. BMC Microbiol. 2020, 20, 226. [Google Scholar] [CrossRef]
- Fattinger, S.A.; Sellin, M.E.; Hardt, W.-D. Epithelial inflammasomes in the defense against Salmonella gut infection. Curr. Opin. Microbiol. 2021, 59, 86–94. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Henry, T. The inflammasomes: Platforms of innate immunity. Med. Sci. 2013, 29, 975–984. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-H.; Rolan, H.G.; Tsolis, R.M. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 2007, 282, 33897–33901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, V.M.R.; Ramirez, J.; Naseer, N.; Palacio, N.M.; Siddarthan, I.J.; Yan, B.M.; Boyer, M.A.; Pensinger, D.A.; Sauer, J.-D.; Shin, S. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2017, 114, 13242–13247. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Rueegg, A.; Werner, S.; Beer, H.-D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1146. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Stecher, B.; Barthel, M.; Kremer, M.; Muller, A.J.; Heikenwalder, M.; Stallmach, T.; Hensel, M.; Pfeffer, K.; Akira, S.; et al. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 2005, 174, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Misaghi, S.; Newton, K.; Maltzman, A.; Izrael-Tomasevic, A.; Arnott, D.; Dixit, V.M. NLRP3 recruitment by NLRC4 during Salmonella infection. J. Exp. Med. 2016, 213, 877–885. [Google Scholar] [CrossRef]
- Storek, K.M.; Monack, D.M. Bacterial recognition pathways that lead to inflammasome activation. Immunol. Rev. 2015, 265, 112–129. [Google Scholar] [CrossRef]
- Kaur, J.; Jain, S.K. Role of antigens and virulence factors of Salmonella enterica serovar Typhi in its pathogenesis. Microbiol. Res. 2012, 167, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Keestra-Gounder, A.M.; Tsolis, R.M.; Baeumler, A.J. Now you see me, now you don’t: The interaction of Salmonella with innate immune receptors. Nat. Rev. Microbiol. 2015, 13, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.-Q.; Song, P.-X.; Chen, W.; Qi, S.; Yu, S.-X.; Du, C.-T.; Deng, X.-M.; Ouyang, H.-S.; Yang, Y.-J. Cirtical role for Salmonella effector SopB in regulating inflammasome activation. Mol. Immunol. 2017, 90, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2396–2401. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.J.; Hoffmann, C.; Galle, M.; Van Den Broeke, A.; Heikenwalder, M.; Falter, L.; Misselwitz, B.; Kremer, M.; Beyaert, R.; Hardt, W.-D. The S. Typhimurium Effector SopE Induces Caspase-1 Activation in Stromal Cells to Initiate Gut Inflammation. Cell Host Microbe 2009, 6, 125–136. [Google Scholar] [CrossRef]
- Bierschenk, D.; Monteleone, M.; Moghaddas, F.; Baker, P.J.; Masters, S.L.; Boucher, D.; Schroder, K. The Salmonella pathogenicity island-2 subverts human NLRP3 and NLRC4 inflammasome responses. J. Leukoc. Biol. 2019, 105, 401–410. [Google Scholar] [CrossRef]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Winter, S.E.; Winter, M.G.; Atluri, V.; Poon, V.; Romao, E.L.; Tsolis, R.M.; Baeumler, A.J. The Flagellar Regulator TviA Reduces Pyroptosis by Salmonella enterica Serovar Typhi. Infect. Immun. 2015, 83, 1546–1555. [Google Scholar] [CrossRef] [Green Version]
- Francis, M.S.; Amer, A.A.; Milton, D.L.; Costa, T.R. Site-Directed Mutagenesis and Its Application in Studying the Interactions of T3S Components. Methods Mol. Biol. 2017, 1531, 11–31. [Google Scholar] [CrossRef]
- O’Toole, G.A.; Kolter, R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: A genetic analysis. Mol. Microbiol. 1998, 28, 449–461. [Google Scholar] [CrossRef]
- Assouvie, A.; Daley-Bauer, L.P.; Rousselet, G. Growing Murine Bone Marrow-Derived Macrophages. Methods Mol. Biol. 2018, 1784, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, F.; Gu, D.; Wang, W.; Huang, J.; Jiao, X. Antimicrobial Effect and the Mechanism of Diallyl Trisulfide against Campylobacter jejuni. Antibiotics 2021, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Newton, K.; Lamkanfi, M.; Mariathasan, S.; Dixit, V.M.; Monack, D.M. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 2010, 207, 1745–1755. [Google Scholar] [CrossRef]
- Bao, H.; Wang, S.; Zhao, J.-H.; Liu, S.L. Salmonella secretion systems: Differential roles in pathogen-host interactions. Microbiol. Res. 2020, 241, 125691. [Google Scholar] [CrossRef]
- Shin, S.; Brodsky, I.E. The inflammasome: Learning from bacterial evasion strategies. Semin. Immunol. 2015, 27, 102–110. [Google Scholar] [CrossRef]
- Philip, N.H.; Zwack, E.E.; Brodsky, I.E. Activation and Evasion of Inflammasomes by Yersinia. Curr. Top Microbiol. Immunol. 2016, 397, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Bierschenk, D.; Boucher, D.; Schroder, K. Salmonella-induced inflammasome activation in humans. Mol. Immunol. 2017, 86, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Fernandez De Henestrosa, A.R.; Badiola, I.; Saco, M.; Perez De Rozas, A.M.; Campoy, S.; Barbe, J. Importance of the galE gene on the virulence of Pasteurella multocida. FEMS Microbiol. Lett. 1997, 154, 311–316. [Google Scholar] [CrossRef]
- Fry, B.N.; Feng, S.; Chen, Y.Y.; Newell, D.G.; Coloe, P.J.; Korolik, V. The galE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Infect. Immun. 2000, 68, 2594–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseiny, M.I.; Hensel, M. Construction of highly attenuated Salmonella enterica serovar Typhimurium live vectors for delivering heterologous antigens by chromosomal integration. Microbiol. Res. 2008, 163, 605–615. [Google Scholar] [CrossRef]
- Gilman, R.H.; Hornick, R.B.; Woodard, W.E.; DuPont, H.L.; Snyder, M.J.; Levine, M.M.; Libonati, J.P. Evaluation of a UDP-glucose-4-epimeraseless mutant of Salmonella typhi as a liver oral vaccine. J. Infect. Dis. 1977, 136, 717–723. [Google Scholar] [CrossRef]
- Clare, B. Inflammasome activation by Salmonella. Curr. Opin. Microbiol. 2021, 64, 27–32. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V. Salmonella and Caspase-1: A complex interplay of detection and evasion. Front Microbiol. 2011, 2, 85. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Lawley, T.D.; Chan, K.; Thompson, L.J.; Kim, C.C.; Govoni, G.R.; Monack, D.M. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006, 2, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Wynosky-Dolfi, M.A.; Snyder, A.G.; Philip, N.H.; Doonan, P.J.; Poffenberger, M.C.; Avizonis, D.; Zwack, E.E.; Riblett, A.M.; Hu, B.; Strowig, T.; et al. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J. Exp. Med. 2014, 211, 653–668. [Google Scholar] [CrossRef] [Green Version]
- Sanman, L.E.; Qian, Y.; Eisle, N.A.; Ng, T.M.; van der Linden, W.A.; Monack, D.M.; Weerapana, E.; Bogyo, M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. eLife 2016, 5, e13663. [Google Scholar] [CrossRef] [PubMed]
- Nnalue, N.A.; Stocker, B.A. Some galE mutants of Salmonella choleraesuis retain virulence. Infect. Immun. 1986, 54, 635–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, N.A.; Ruby, T.; Jacobson, A.; Manzanillo, P.S.; Cox, J.S.; Lam, L.; Mukundan, L.; Chawla, A.; Monack, D.M. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 2013, 14, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giblin, J.P.; Hewlett, L.J.; Hannah, M.J. Basal secretion of von Willebrand factor from human endothelial cells. Blood 2008, 112, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Gu, J.; Zhu, J.; Wang, X.; Wang, C.; Duan, C.; Ni, Y.; Lu, X.; Li, J. Up-regulation of Dyrk1b promote astrocyte activation following lipopolysaccharide-induced neuroinflammation. Neuropeptides 2018, 69, 76–83. [Google Scholar] [CrossRef]
- Gervais, O.; Ueno, K.; Kawai, Y.; Hitomi, Y.; Aiba, Y.; Ueta, M.; Nakamura, M.; Tokunaga, K.; Nagasaki, M. Regional heritability mapping identifies several novel loci (STAT4, ULK4, and KCNH5) for primary biliary cholangitis in the Japanese population. Eur. J. Hum. Genet. 2021, 29, 1282–1291. [Google Scholar] [CrossRef]
- Wang, M.; Jing, J.; Li, H.; Liu, J.; Yuan, Y.; Sun, L. The expression characteristics and prognostic roles of autophagy-related genes in gastric cancer. PeerJ 2021, 9, e10814. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ma, J.; Li, D.; Li, P.; Zhou, X.; Li, Y.; He, Z.; Qin, L.; Liang, L.; Luo, X. Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. J. Neuroinflamm. 2019, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Xia, Z.; Zhou, X.; Guo, Y.; Guo, R.; Kang, X.; Wu, K.; Sun, J.; Xu, X.; Jiao, X.; et al. Signature-tagged mutagenesis screening revealed the role of lipopolysaccharide biosynthesis gene rfbH in smooth-to-rough transition in Salmonella Enteritidis. Microbiol. Res. 2018, 212–213, 75–79. [Google Scholar] [CrossRef]
- Chiang, S.L.; Rubin, E.J. Construction of a mariner-based transposon for epitope-tagging and genomic targeting. Gene 2002, 296, 179–185. [Google Scholar] [CrossRef]
- Wang, S.Y.; Lauritz, J.; Jass, J.; Milton, D.L. A ToxR homolog from Vibrio anguillarum serotype O1 regulates its own production, bile resistance, and biofilm formation. J. Bacteriol. 2002, 184, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, X.; Oswald, E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J. Bacteriol. 2004, 186, 5486–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.; Gu, D.; Guo, Y.; Li, A.; Kang, X.; Jiao, X.; Pan, Z. Salmonella Enteritidis GalE Protein Inhibits LPS-Induced NLRP3 Inflammasome Activation. Microorganisms 2022, 10, 911. https://doi.org/10.3390/microorganisms10050911
Huang T, Gu D, Guo Y, Li A, Kang X, Jiao X, Pan Z. Salmonella Enteritidis GalE Protein Inhibits LPS-Induced NLRP3 Inflammasome Activation. Microorganisms. 2022; 10(5):911. https://doi.org/10.3390/microorganisms10050911
Chicago/Turabian StyleHuang, Tingting, Dan Gu, Yaxin Guo, Ang Li, Xilong Kang, Xinan Jiao, and Zhiming Pan. 2022. "Salmonella Enteritidis GalE Protein Inhibits LPS-Induced NLRP3 Inflammasome Activation" Microorganisms 10, no. 5: 911. https://doi.org/10.3390/microorganisms10050911
APA StyleHuang, T., Gu, D., Guo, Y., Li, A., Kang, X., Jiao, X., & Pan, Z. (2022). Salmonella Enteritidis GalE Protein Inhibits LPS-Induced NLRP3 Inflammasome Activation. Microorganisms, 10(5), 911. https://doi.org/10.3390/microorganisms10050911