
Fecal Microbiome Differences in Angus Steers with Differing Feed Efficiencies during the Feedlot-Finishing Phase

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sample Collection, DNA Extraction, and Sequencing
2.3. 16S rRNA Gene Sequencing Data Analysis
2.4. Statistical Analysis
3. Results
3.1. Animal Performance
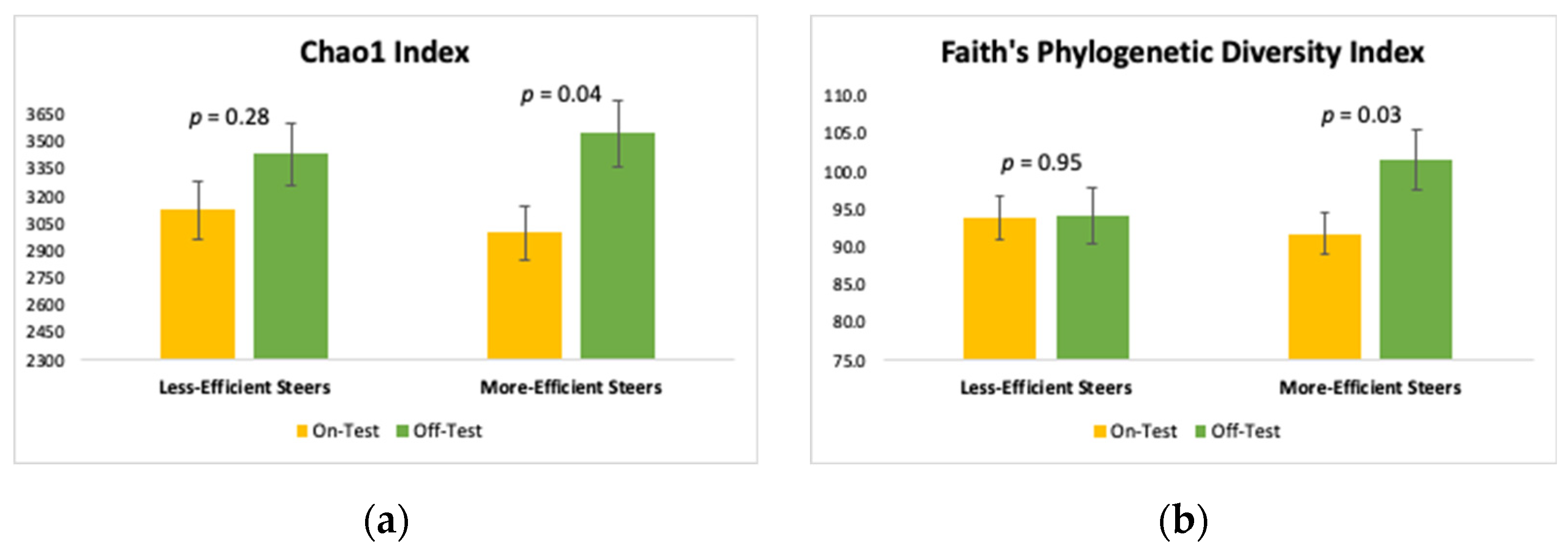
3.2. Alpha Diversity
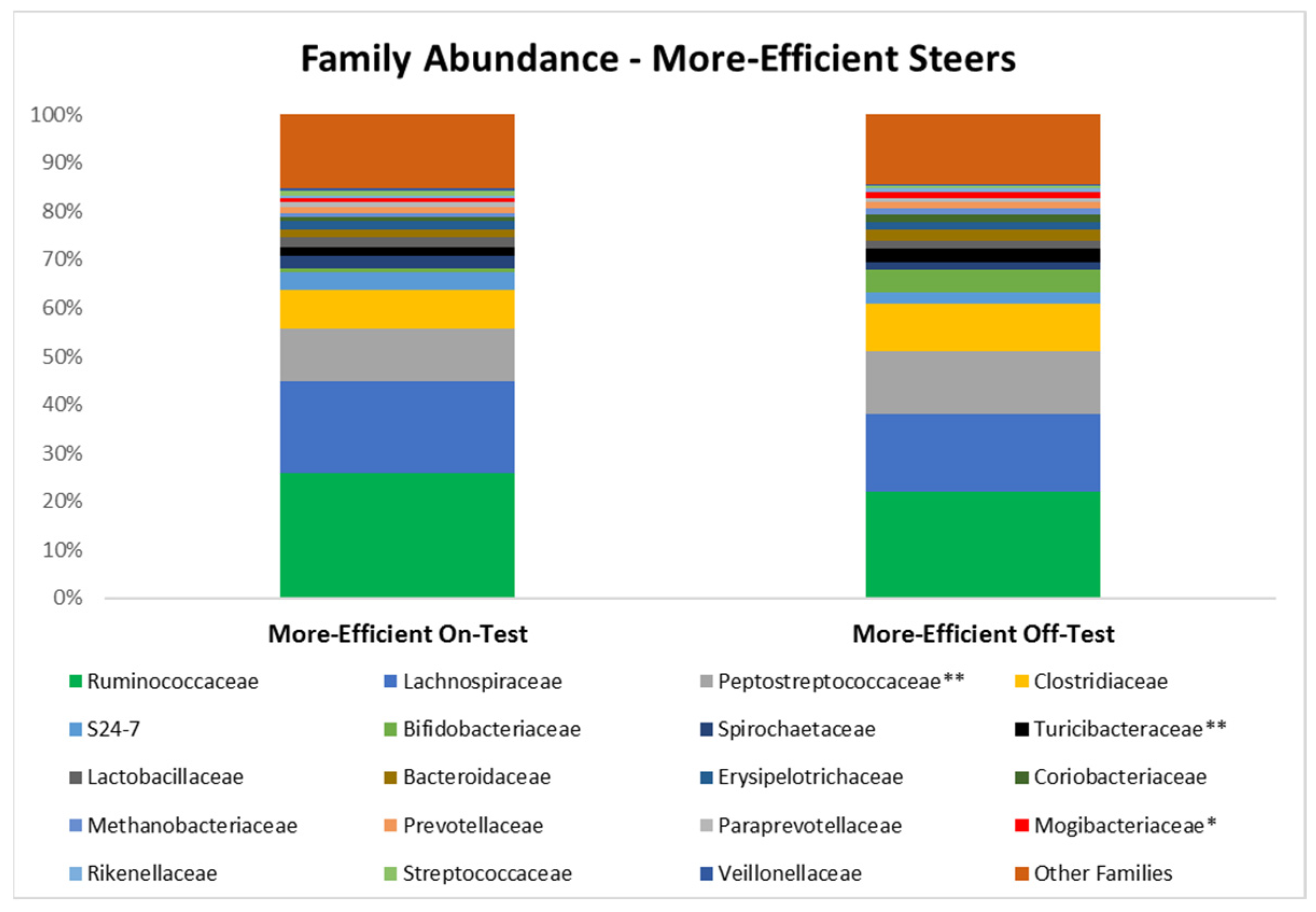
3.3. Microbial Taxa
4. Discussion
4.1. Animal Performance
4.2. Changes in Microbial Diversity
4.3. Changes in Specific Microbial Taxa in the Hindgut
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McAnally, A.R.; Phillipson, A.T. Digestion in the ruminant. Biol. Rev. 1944, 19, 41–54. [Google Scholar] [CrossRef]
- Carberry, C.A.; Kenny, D.A.; Han, S.; McCabe, M.S.; Waters, S.M. Effect of phenotypic residual feed intake and dietary forage content on the rumen microbial community of beef cattle. Appl. Env. Microbiol. 2012, 78, 4949–4958. [Google Scholar] [CrossRef] [PubMed]
- Shabat, S.K.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guan, L.L. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef] [PubMed]
- McGovern, E.; McGee, M.; Byrne, C.J.; Kenny, D.A.; Kelly, A.K.; Waters, S.M. Investigation into the effect of divergent feed efficiency phenotype on the bovine rumen microbiota across diet and breed. Sci. Rep. 2020, 10, 15317. [Google Scholar] [CrossRef]
- Boaitey, A.; Goddard, E.; Mohapatra, S.; Crowley, J. Feed efficiency estimates in cattle: The economic and environmental impacts of reranking. Sustain. Agric. Res. 2017, 6, 35–47. [Google Scholar] [CrossRef]
- Gilbert, H.; Bidanel, J.P.; Gruand, J.; Caritez, J.C.; Billon, Y.; Guillouet, P. Genetic parameters for residual feed intake in growing pigs, with emphasis on genetic relationships with carcass and meat quality traits. J. Anim. Sci. 2007, 85, 3182–3188. [Google Scholar] [CrossRef]
- Herd, R.M.; Arthur, P.F. Physiological basis for residual feed intake. J. Anim. Sci. 2009, 87, E64–E71. [Google Scholar] [CrossRef]
- Chen, Y.; Gondro, C.; Quinn, K.; Herd, R.M.; Parnell, P.F.; Vanselow, B. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim. Genet. 2011, 42, 475–490. [Google Scholar] [CrossRef]
- Krause, D.O.; Nagaraja, T.G.; Wright, A.D.G.; Callaway, T.R. Rumen microbiology: Leading the way in microbial ecology. J. Anim. Sci. 2013, 91, 331–341. [Google Scholar] [CrossRef]
- Hernandez-Sanabria, E.; Goonewardene, L.A.; Wang, Z.; Durunna, O.N.; Moore, S.S.; Guan, L.L. Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl. Environ. Microbiol. 2012, 78, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Myer, P.R.; Smith, T.P.L.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C. Rumen Microbiome from Steers Differing in Feed Efficiency. PLoS ONE 2015, 10, e0129174. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.J.; Huws, S.A.; Edwards, J.E.; Kingston-Smith, A.H.; Siu-Ting, K.; Hughes, M.; Rubino, F.; Friedersdorff, M.; Creevey, C.J. CowPI: A rumen microbiome focussed version of the PICRUSt functional inference software. Front. Microbiol. 2018, 9, 1095. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2014, 116, 245–257. [Google Scholar] [CrossRef]
- Lourenco, J.M.; Callaway, T.R.; Kieran, T.J.; Glenn, T.C.; McCann, J.C.; Stewart, R.L., Jr. Analysis of the Rumen Microbiota of Beef Calves Supplemented during the Suckling Phase. Front. Microbiol. 2019, 10, 1131. [Google Scholar] [CrossRef] [PubMed]
- Myer, P.R.; Wells, J.E.; Smith, T.P.; Kuehn, L.A.; Freetly, H.C. Cecum microbial communities from steers differing in feed efficiency. J. Anim. Sci. 2015, 93, 5327–5340. [Google Scholar] [CrossRef]
- Welch, C.B.; Lourenco, J.M.; Davis, D.B.; Krause, T.R.; Carmichael, M.N.; Rothrock, M.J.; Pringle, T.D.; Callaway, T.R. The impact of feed efficiency selection on the ruminal, cecal, and fecal microbiomes of Angus steers from a commercial feedlot. J. Anim. Sci. 2020, 98, 7. [Google Scholar] [CrossRef]
- Welch, C.B.; Lourenco, J.M.; Krause, T.R.; Seidel, D.S.; Fluharty, F.L.; Pringle, T.D.; Callaway, T.R. Evaluation of the Fecal Bacterial Communities of Angus Steers with Divergent Feed Efficiencies Across the Lifespan from Weaning to Slaughter. Front. Vet. Sci. 2021, 8, 597405. [Google Scholar] [CrossRef]
- Detweiler, R.A.; Pringle, T.D.; Rekaya, R.; Wells, J.B.; Segers, J.R. The impact of selection using residual average daily gain and marbling EPDs on growth, performance, and carcass traits in Angus steers. J. Anim. Sci. 2019, 97, 2450–2459. [Google Scholar] [CrossRef]
- NRC. Nutrient Requirements of Beef Cattle: Seventh Revised Edition: Update 2000; The National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Rothrock, M.J., Jr.; Hiett, K.L.; Gamble, J.; Caudill, A.C.; Cicconi-Hogan, K.M.; Caporaso, J.G. A hybrid DNA extraction method for the qualitative and quantitative assessment of bacterial communities from poultry production samples. J. Vis. Exp. 2014, 94, e52161. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, J.M.; Kieran, T.J.; Seidel, D.S.; Glenn, T.C.; da Silveira, M.F.; Callaway, T.R.; Stewart, R.L., Jr. Comparison of the ruminal and fecal microbiotas in beef calves supplemented or not with concentrate. PLoS ONE 2020, 15, e0231533. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Welch, C.B.; Lourenco, J.M.; Seidel, D.S.; Krause, T.R.; Rothrock, M.J.; Pringle, T.D.; Callaway, T.R. The Impact of Pre-Slaughter Fasting on the Ruminal Microbial Population of Commercial Angus Steers. Microorganisms 2021, 9, 2625. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, R.S.; Goopy, J.P.; Herd, R.M.; McCorkell, B. Cattle selected for lower residual feed intake have reduced daily methane production. J. Anim. Sci. 2007, 85, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wofford, H.; Holechek, J.L.; Galyean, M.L.; Wallace, J.D.; Cardenas, M. Evaluation of fecal indices to predict cattle diet quality. J. Range Manag. 1985, 38, 450–454. [Google Scholar] [CrossRef]
- Sezenna, M.L. Proteobacteria: Phylogeny, Metabolic Diversity and Ecological Effects; Nova Science Publishers, Inc.: New York, NY, USA, 2011. [Google Scholar]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A common factor in human diseases. BioMed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef]
- Slobodkin, A. The family Peptostreptococcaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Russell, J.B.; O’connor, J.D.; Fox, D.G.; van Soest, P.J.; Sniffen, C.J. A net carbohydrate and protein system for evaluating cattle diets: I. Ruminal fermentation. J. Anim. Sci. 1992, 70, 3551–3561. [Google Scholar] [CrossRef]
- Fu, L.; Qiu, Y.; Shen, L.; Cui, C.; Wang, S.; Wang, S.; Xie, Y.; Zhao, X.; Gao, X.; Ning, G.; et al. The delayed effects of antibiotics in type 2 diabetes, friend or foe? J. Endocrinol. 2018, 238, 137–149. [Google Scholar] [CrossRef]
- Abbas, W.; Knoell, A.L.; Tom, W.A.; Anderson, C.L.; Paz, H.A.; Fernando, S.C. Impact of Rumen Bacteria on Marbling in Wagyu Cattle. J. Anim. Sci. 2018, 96, 245–246. [Google Scholar] [CrossRef][Green Version]
- Tang, M.T.; Han, H.; Yu, Z.; Tsuruta, T.; Nishino, N. Variability, stability, and resilience of fecal microbiota in dairy cows fed whole crop corn silage. Appl. Microbiol. Biotechnol. 2017, 101, 6355–6364. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.C.; Maxwell, C.V.; Zhao, J. Lifelong dynamics of the swine gut microbiome: From birth to market. J. Anim. Sci. 2019, 97, 48. [Google Scholar] [CrossRef]
- Henning, S.M.; Yang, J.; Hsu, M.; Lee, R.P.; Grojean, E.M.; Ly, A.; Tseng, C.H.; Heber, D.; Li, Z. Decaffeinated green and black tea polyphenols decrease weight gain and alter microbiome populations and function in diet-induced obese mice. Eur. J. Nutr. 2018, 57, 2759–2769. [Google Scholar] [CrossRef]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria–From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef]
- Konikoff, T.; Gophna, U. Oscillospira: A Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol. 2016, 24, 523–524. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Item | Steer Classification | p-Value 1 | |
|---|---|---|---|
| Inefficient (High RFI) | Efficient (Low RFI) | ||
| Average daily gain, kg/day | 1.05 | 1.02 | 0.82 |
| Dry matter intake, kg/day | 13.02 | 10.89 | 0.03 |
| Residual Feed Intake, kg/day | 0.76 | −1.09 | 0.003 |
| Hot carcass weight, kg | 367.9 | 378.6 | 0.62 |
| Taxa | Abundance during Feedlot | p-Value | |
|---|---|---|---|
| Beginning (%) | End (%) | ||
| Family Ruminococcaceae | 24.01 | 18.35 | 0.02 |
| Family Clostridiaceae | 8.49 | 12.76 | 0.01 |
| Family Veillonellaceae | 0.34 | 0.63 | 0.07 |
| Genus Clostridium | 0.86 | 1.12 | 0.01 |
| Genus Oscillospira | 0.53 | 0.83 | 0.04 |
| Taxa | Abundance during Feedlot | p-Value | |
|---|---|---|---|
| Beginning (%) | End (%) | ||
| Phylum Proteobacteria | 0.80 | 0.36 | 0.096 |
| Family Peptostreptococcaceae | 10.75 | 12.94 | 0.03 |
| Family Turicibacteraceae | 1.59 | 2.81 | 0.01 |
| Family Mogibacteriaceae | 0.63 | 1.17 | 0.08 |
| Genus Turicibacter | 1.59 | 2.81 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenco, J.M.; Welch, C.B.; Krause, T.R.; Wieczorek, M.A.; Fluharty, F.L.; Rothrock, M.J.; Pringle, T.D.; Callaway, T.R. Fecal Microbiome Differences in Angus Steers with Differing Feed Efficiencies during the Feedlot-Finishing Phase. Microorganisms 2022, 10, 1128. https://doi.org/10.3390/microorganisms10061128
Lourenco JM, Welch CB, Krause TR, Wieczorek MA, Fluharty FL, Rothrock MJ, Pringle TD, Callaway TR. Fecal Microbiome Differences in Angus Steers with Differing Feed Efficiencies during the Feedlot-Finishing Phase. Microorganisms. 2022; 10(6):1128. https://doi.org/10.3390/microorganisms10061128
Chicago/Turabian StyleLourenco, Jeferson M., Christina B. Welch, Taylor R. Krause, Michael A. Wieczorek, Francis L. Fluharty, Michael J. Rothrock, T. Dean Pringle, and Todd R. Callaway. 2022. "Fecal Microbiome Differences in Angus Steers with Differing Feed Efficiencies during the Feedlot-Finishing Phase" Microorganisms 10, no. 6: 1128. https://doi.org/10.3390/microorganisms10061128
APA StyleLourenco, J. M., Welch, C. B., Krause, T. R., Wieczorek, M. A., Fluharty, F. L., Rothrock, M. J., Pringle, T. D., & Callaway, T. R. (2022). Fecal Microbiome Differences in Angus Steers with Differing Feed Efficiencies during the Feedlot-Finishing Phase. Microorganisms, 10(6), 1128. https://doi.org/10.3390/microorganisms10061128