
Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors

 , , and
, , and
Abstract
:1. Introduction
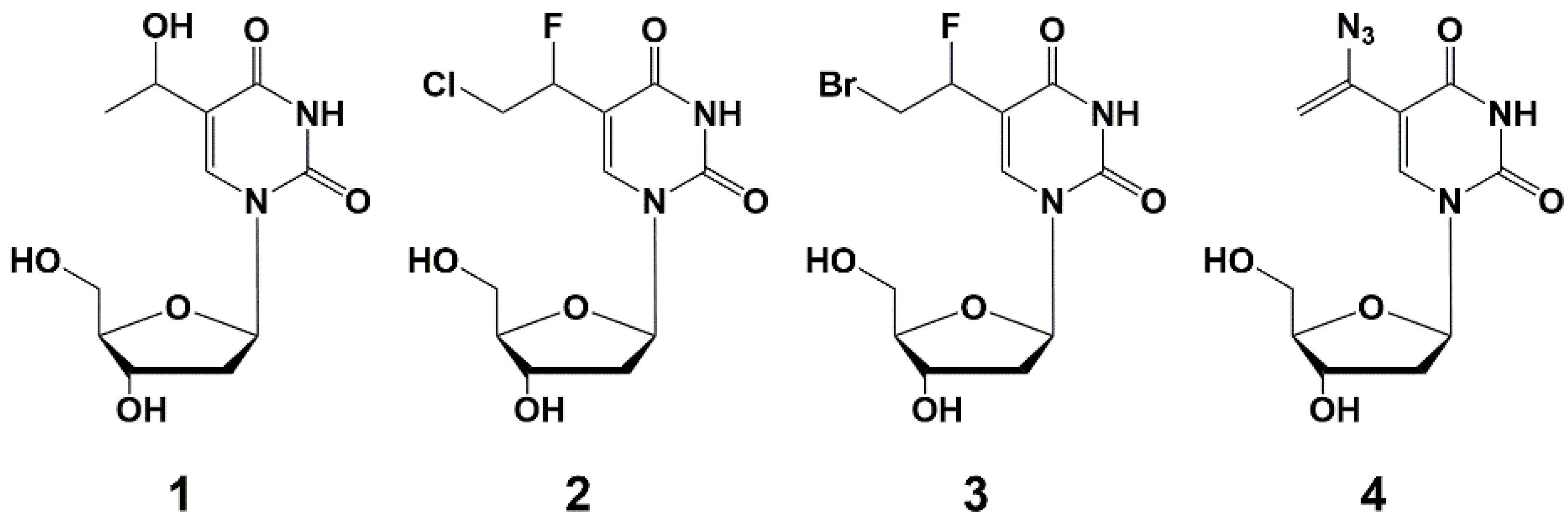
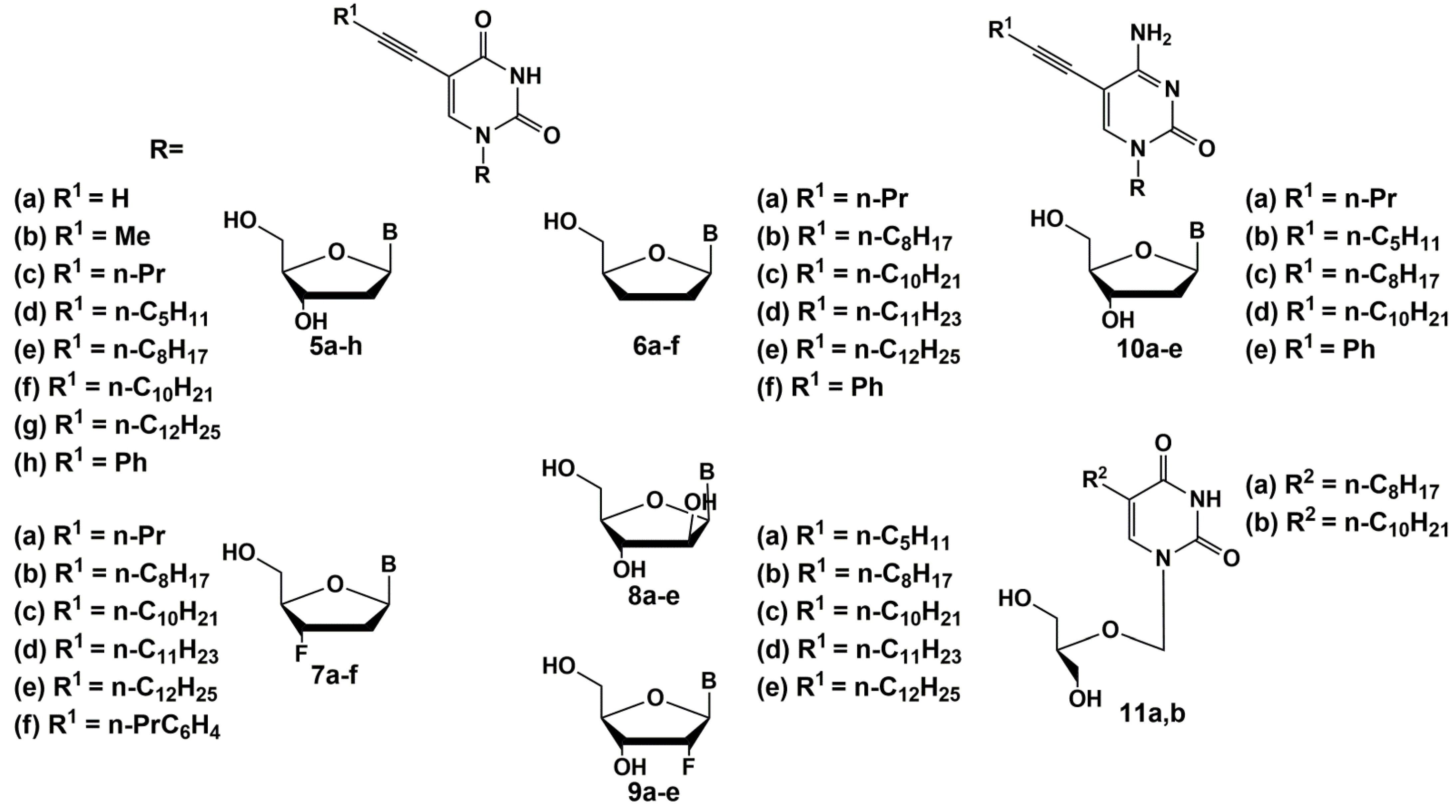
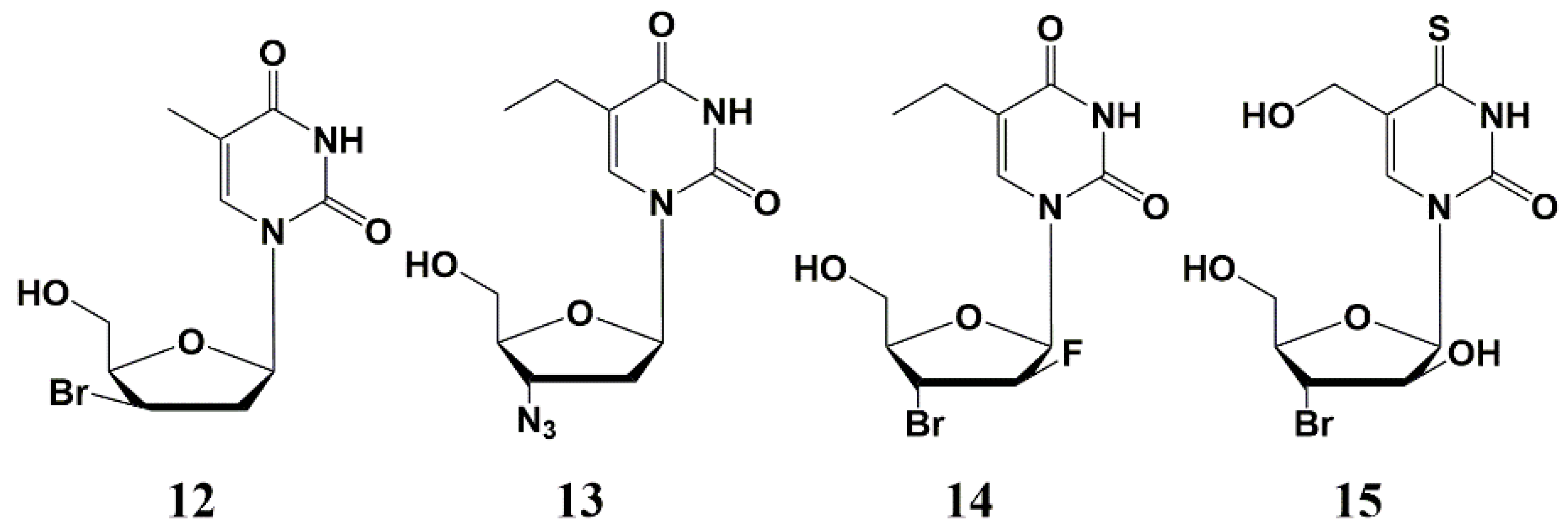
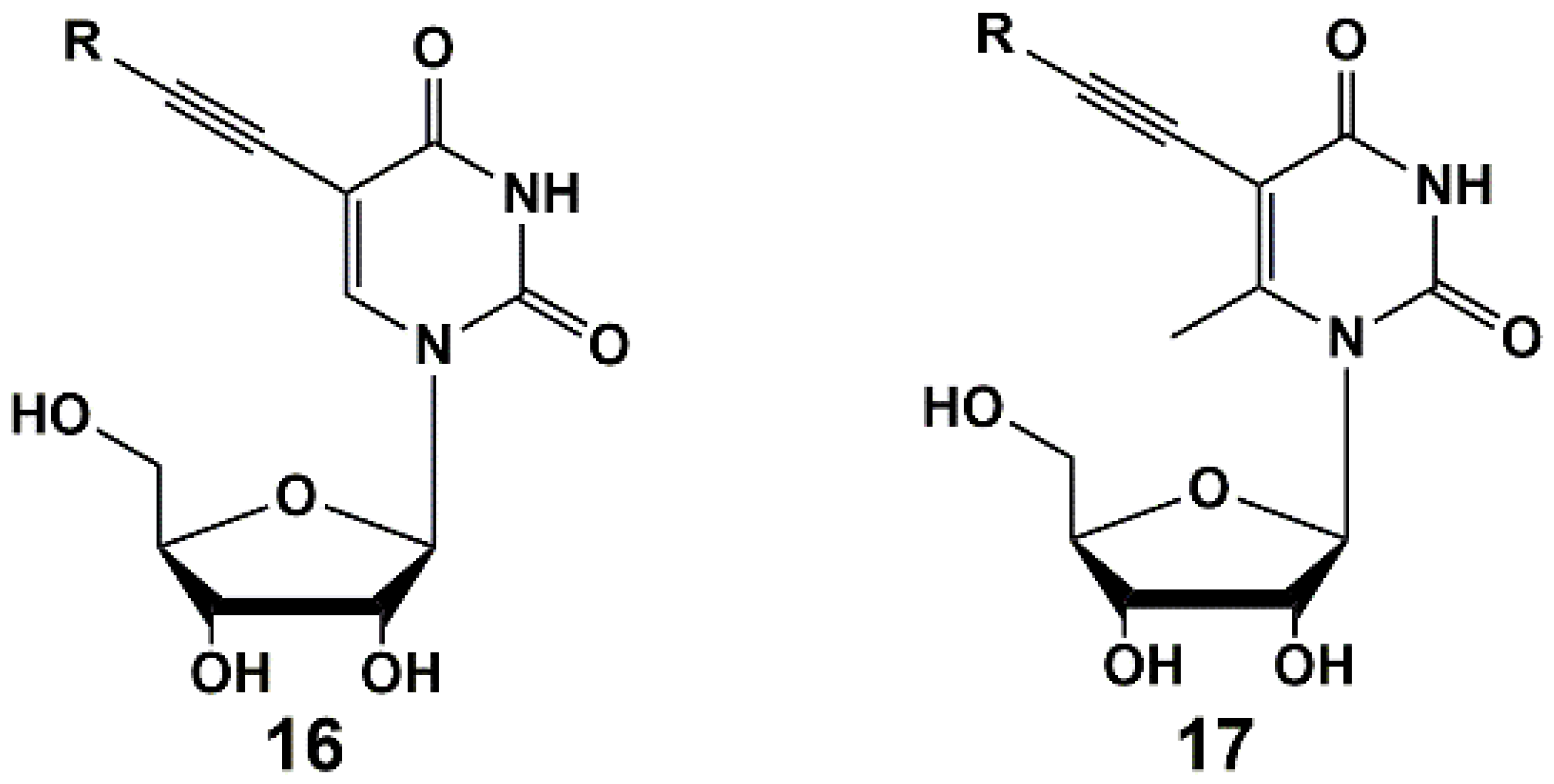
2. Inhibitors of M. tuberculosis Growth with Unidentified Target
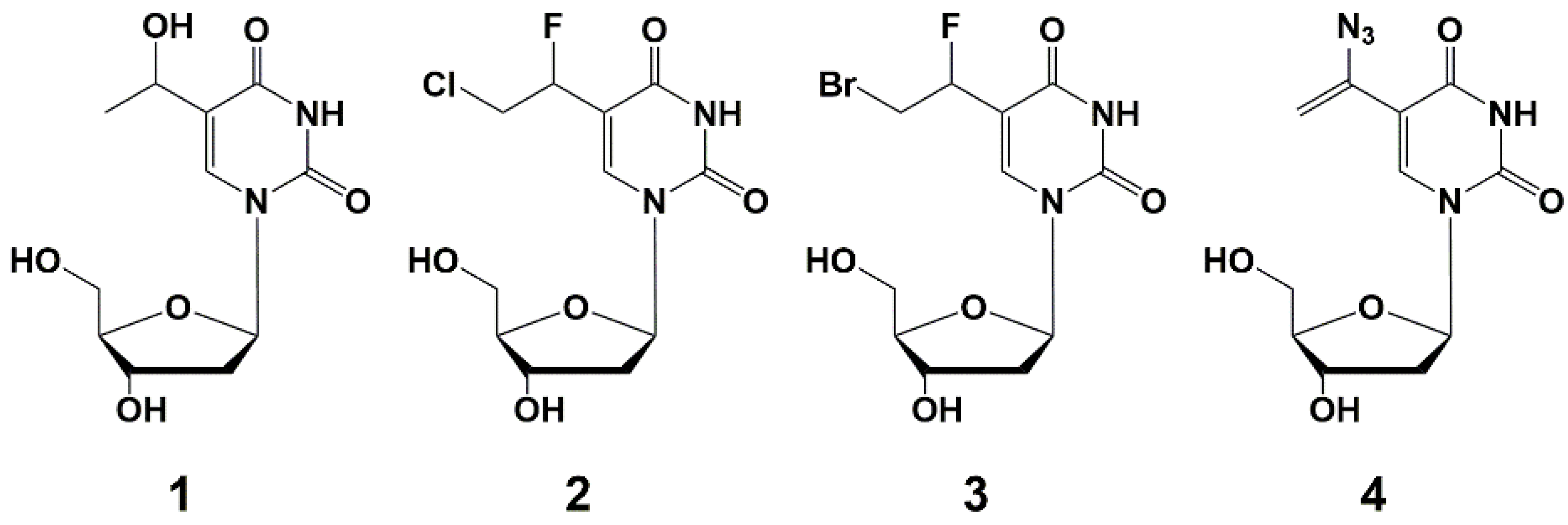
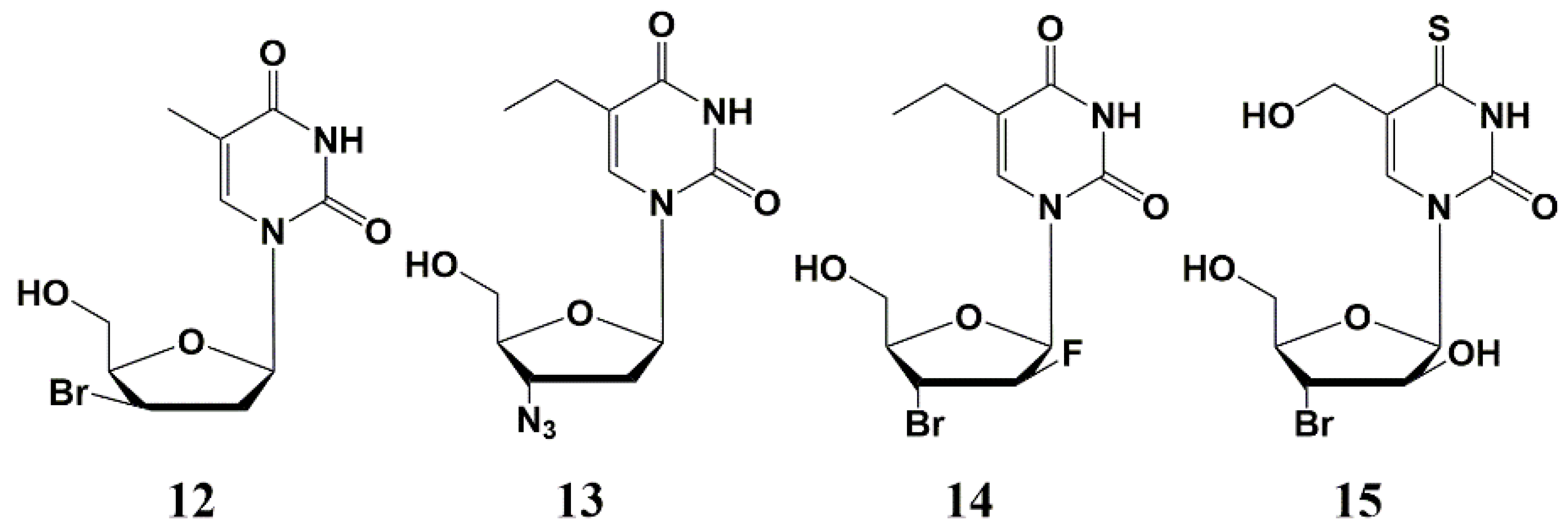
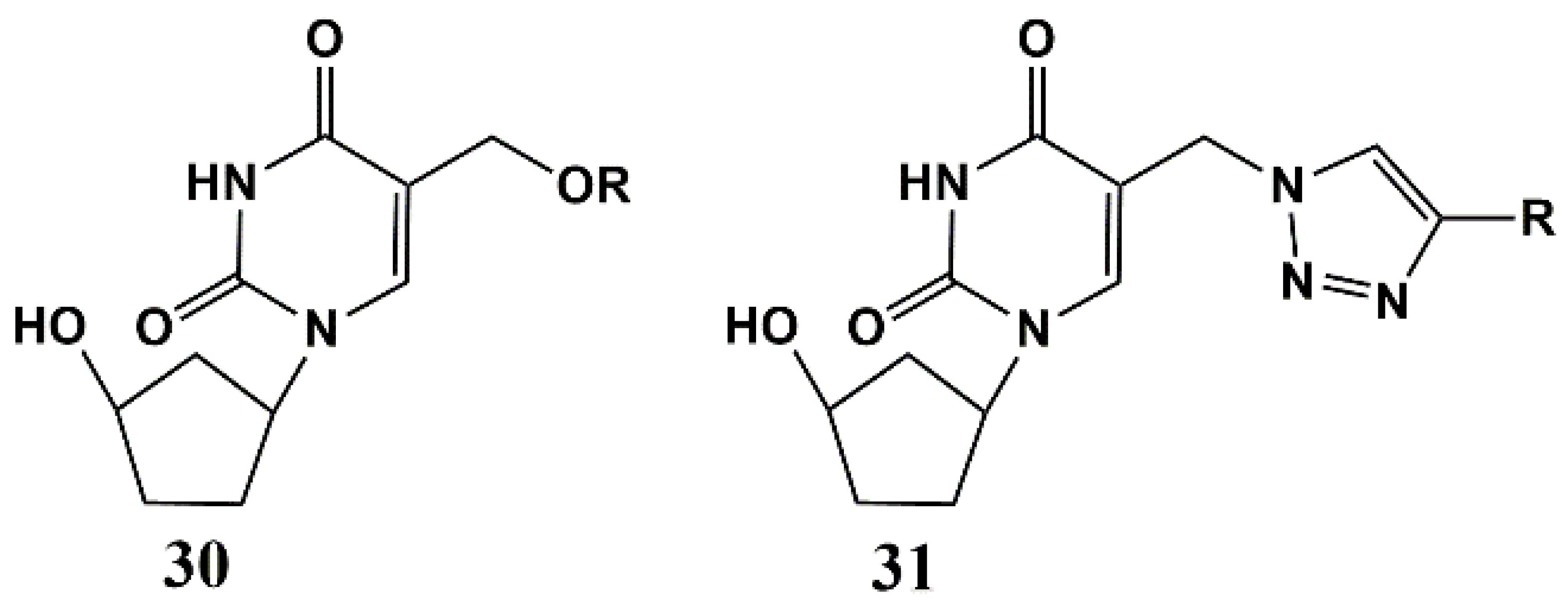
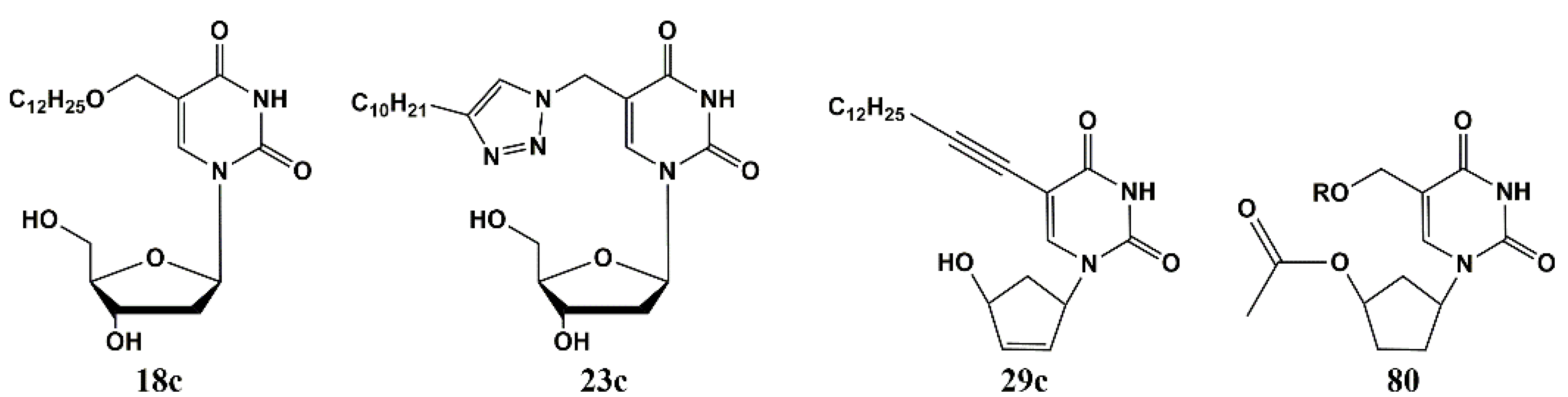
5′-Norcarbocyclic Uracil Derivatives
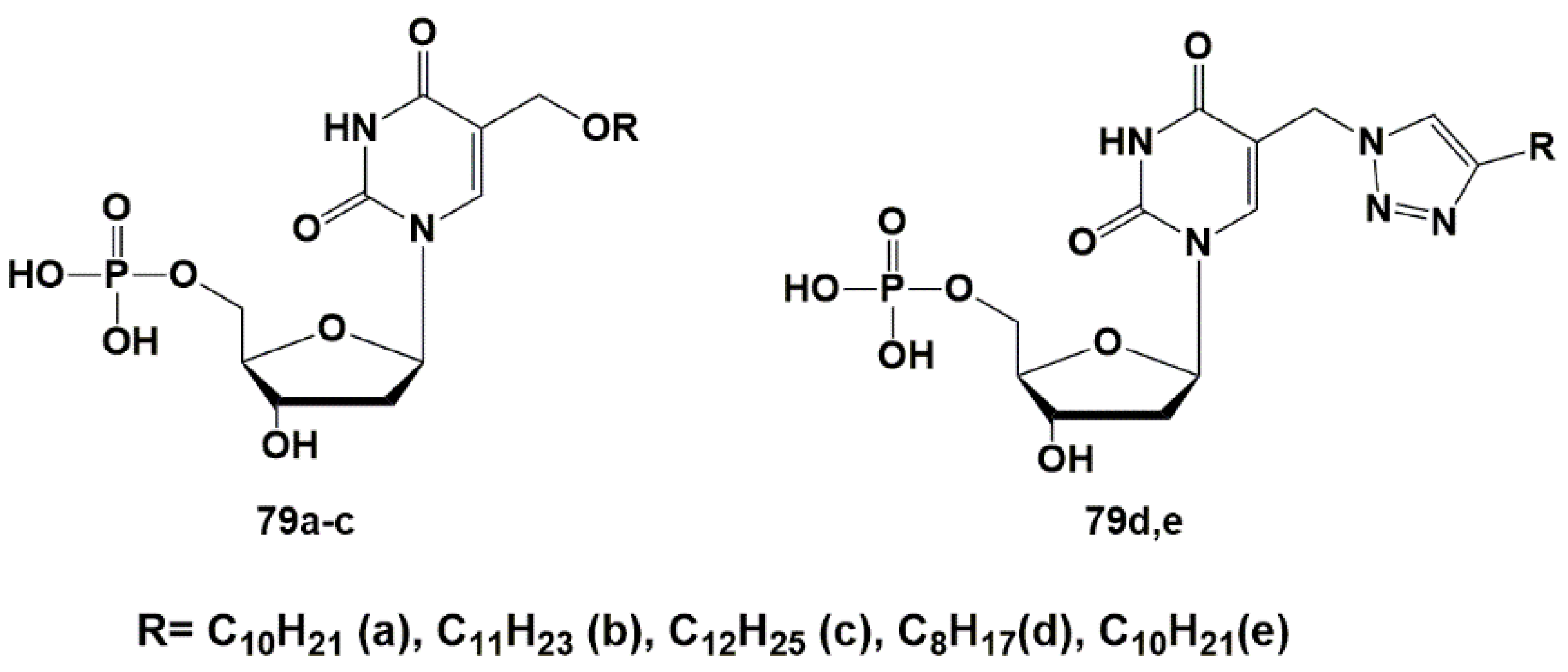
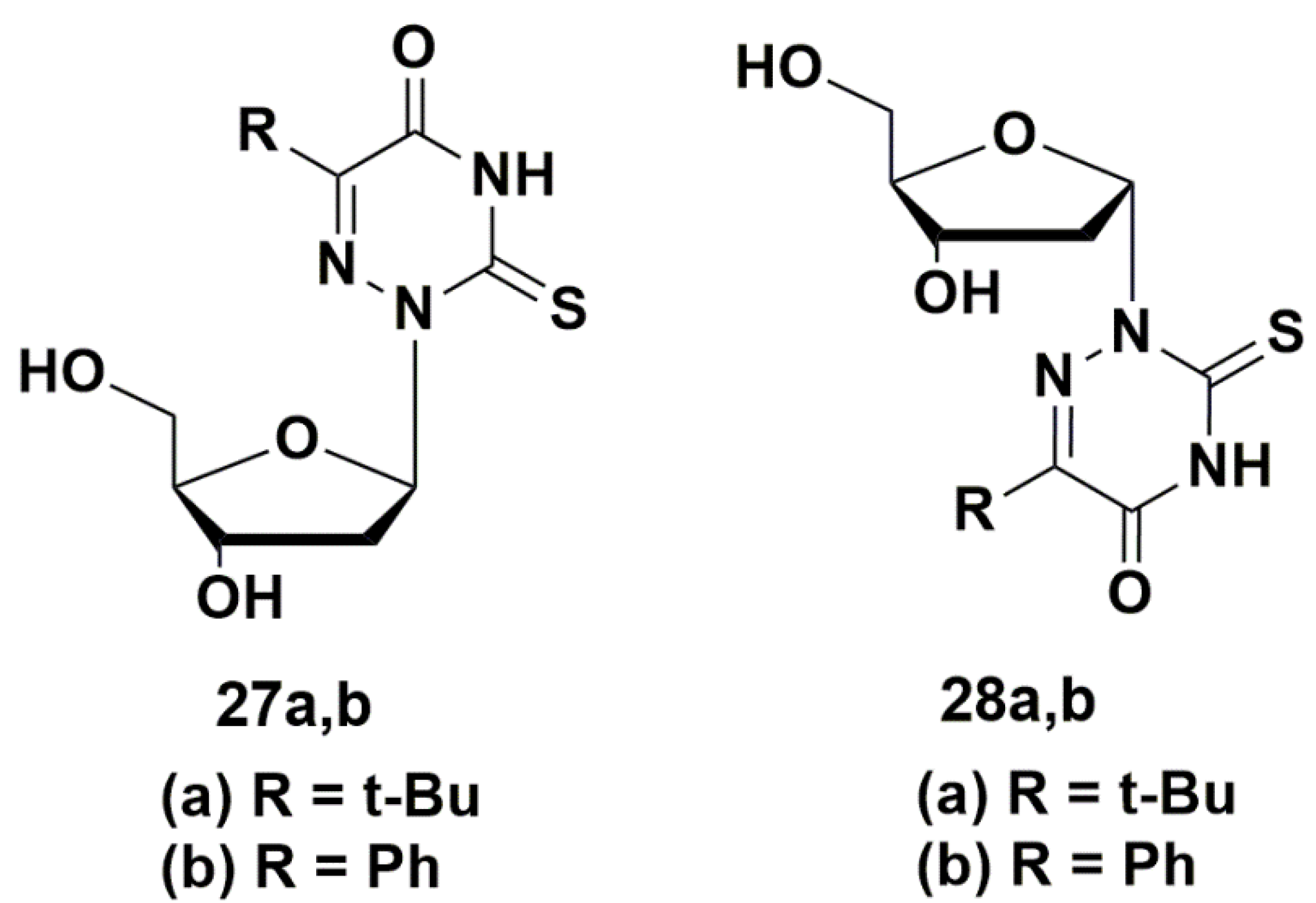
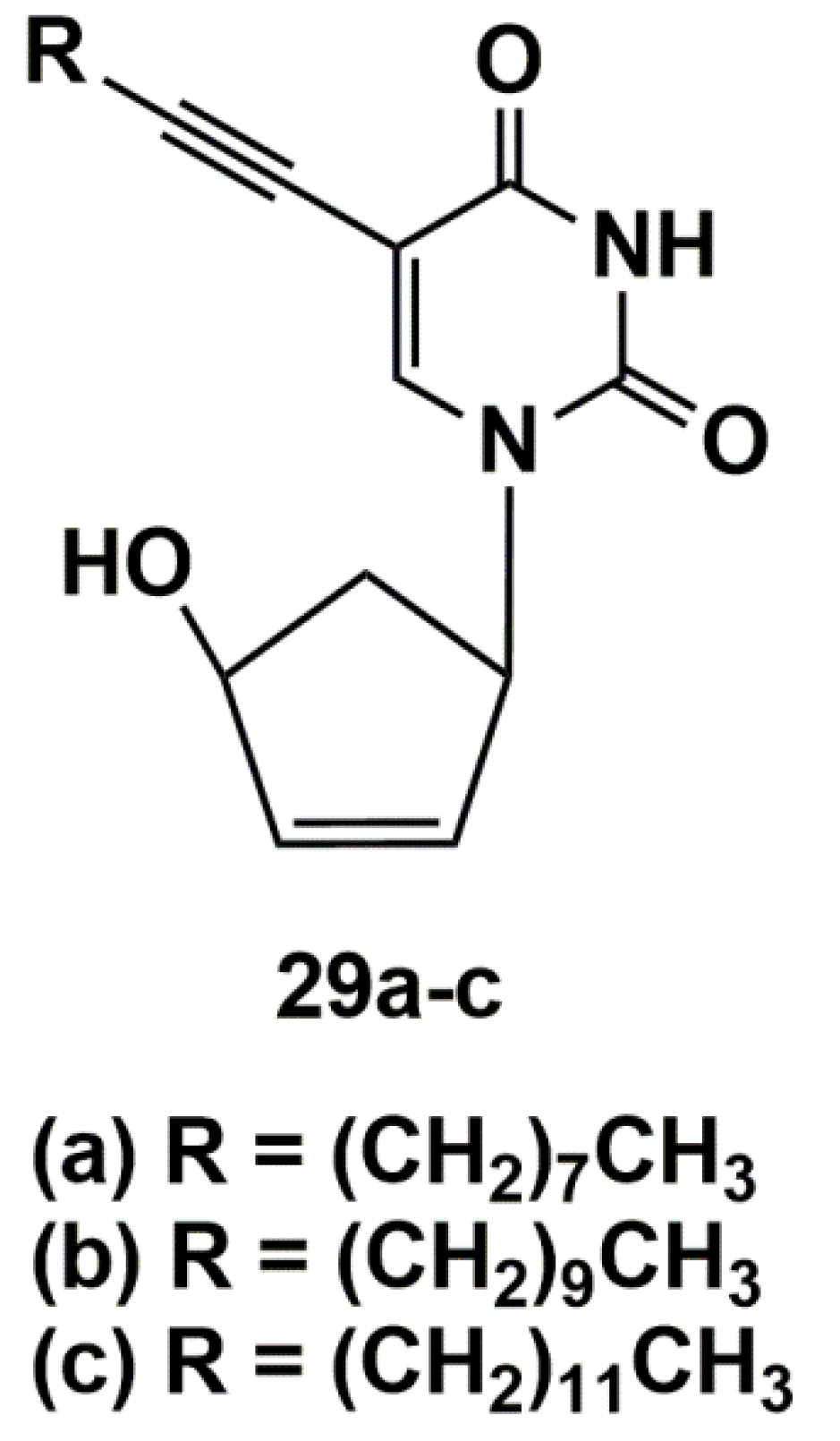
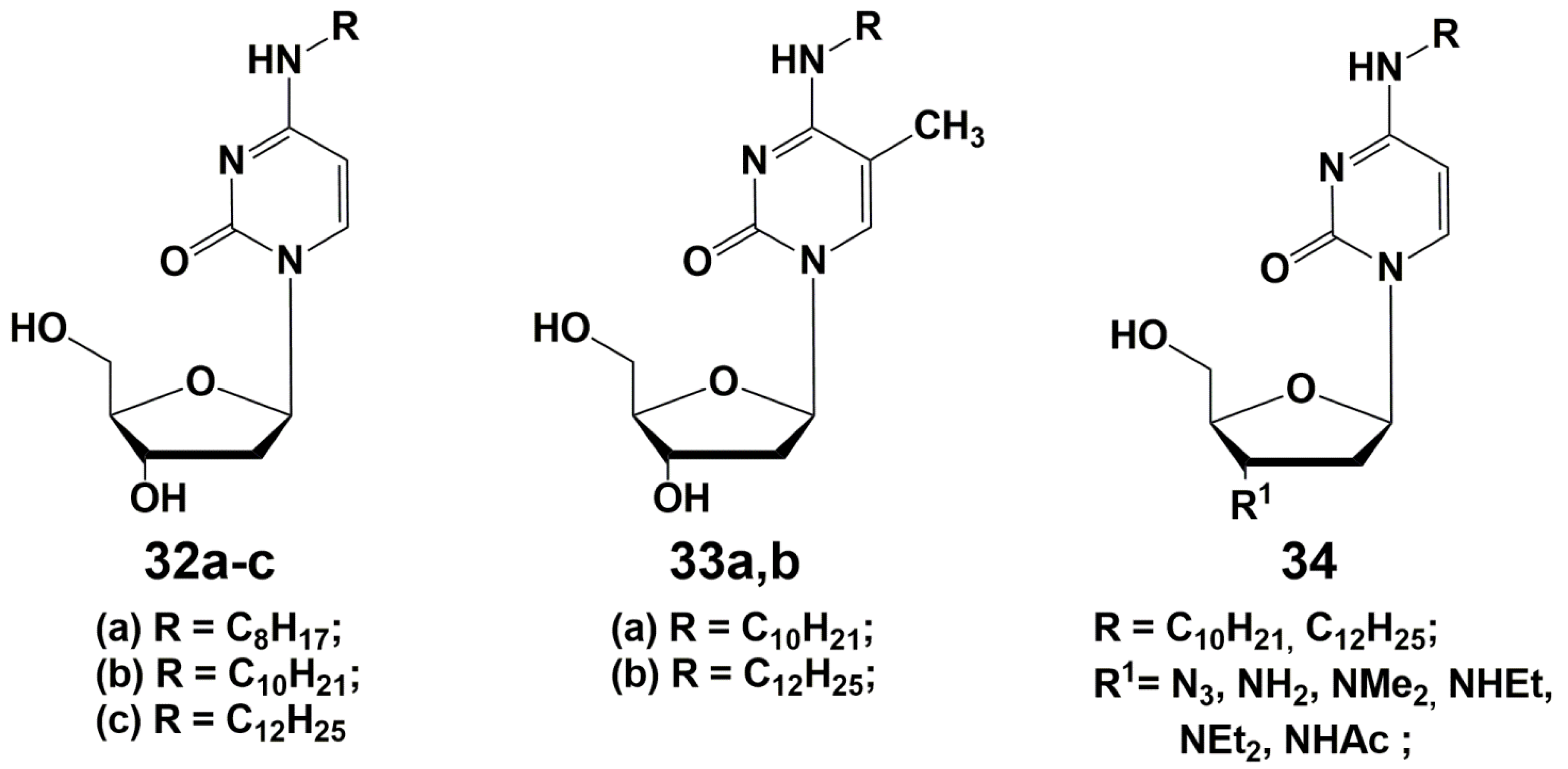
3. N4-Modified Cytidine Derivatives
4. Depot Forms
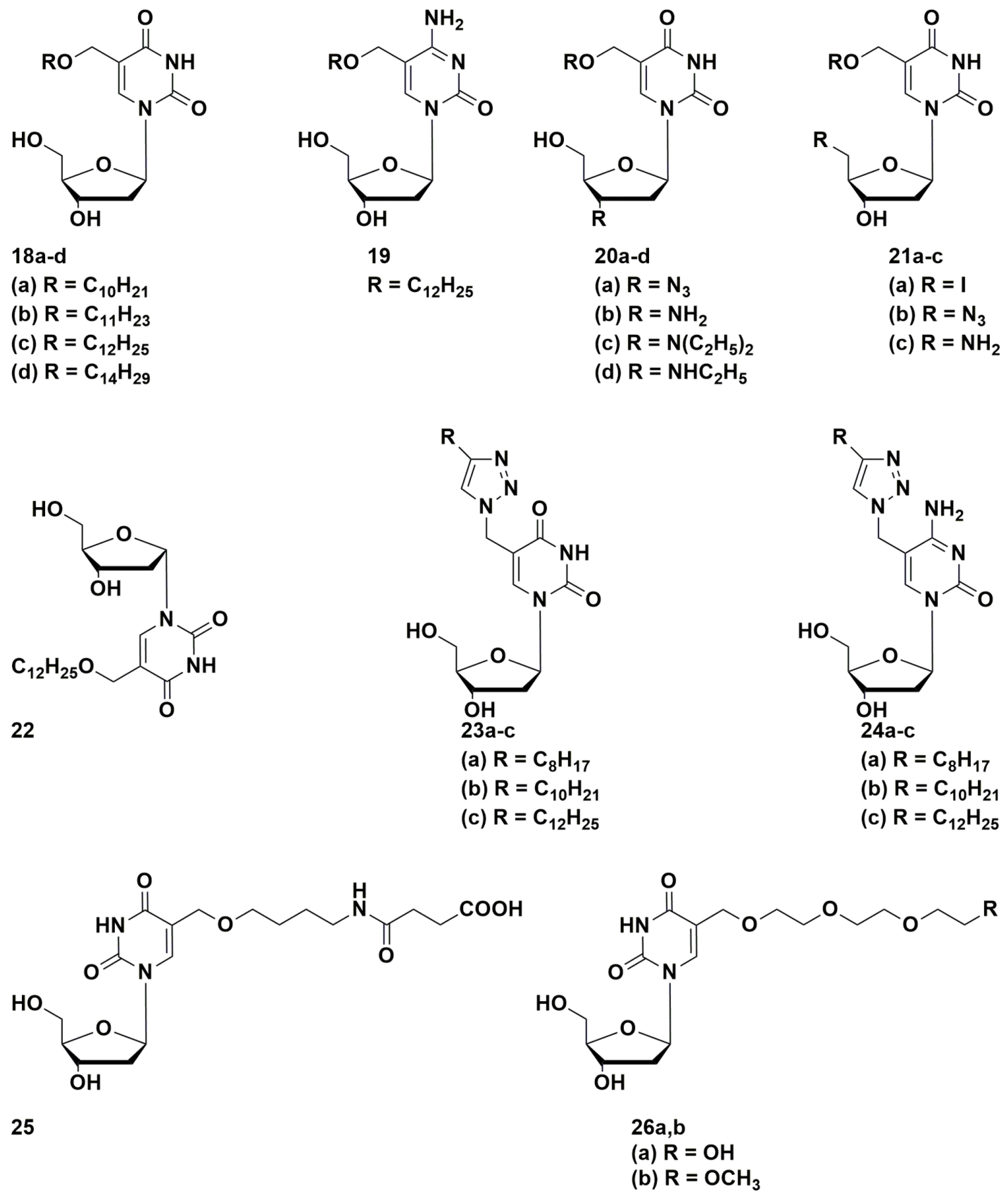
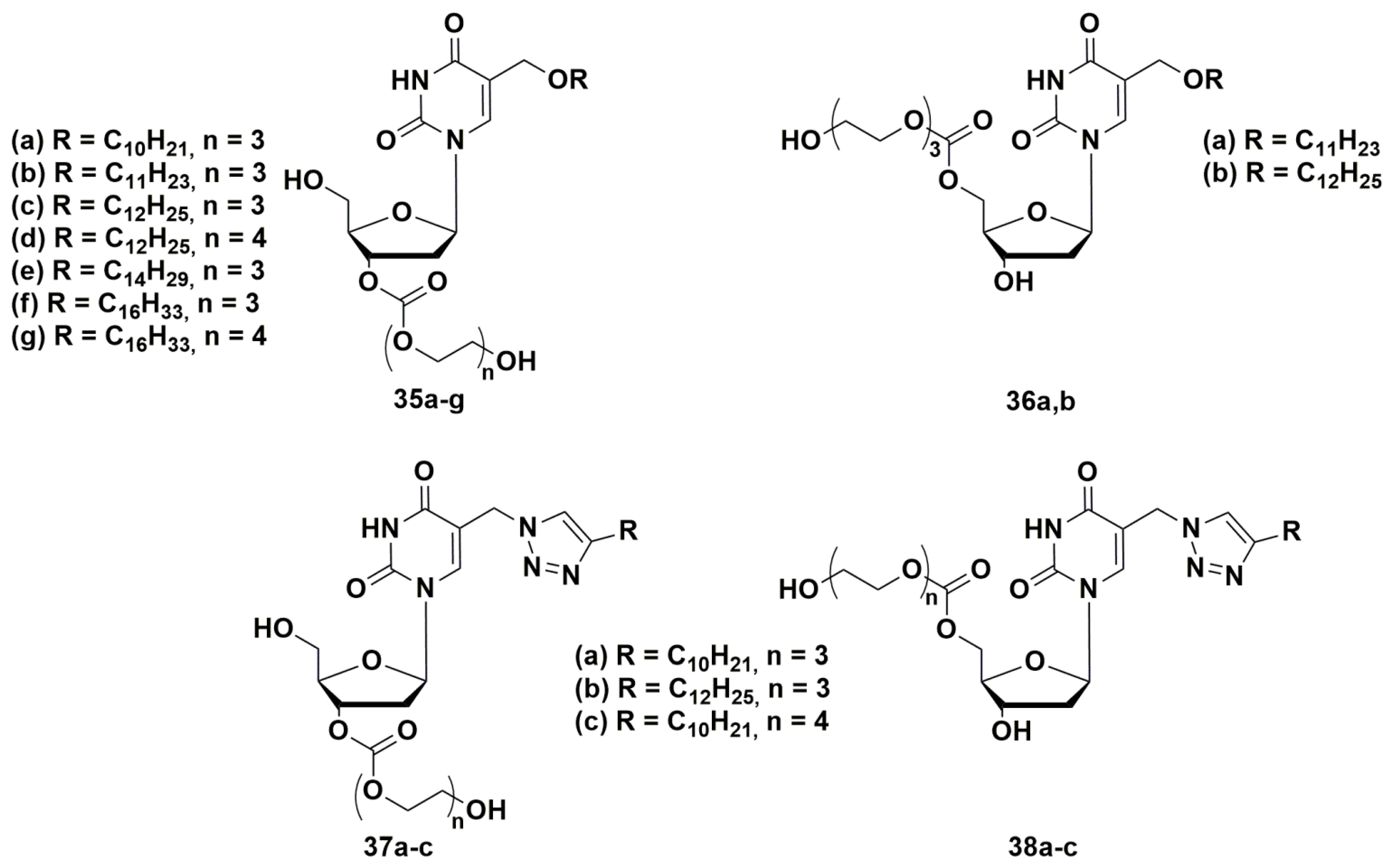
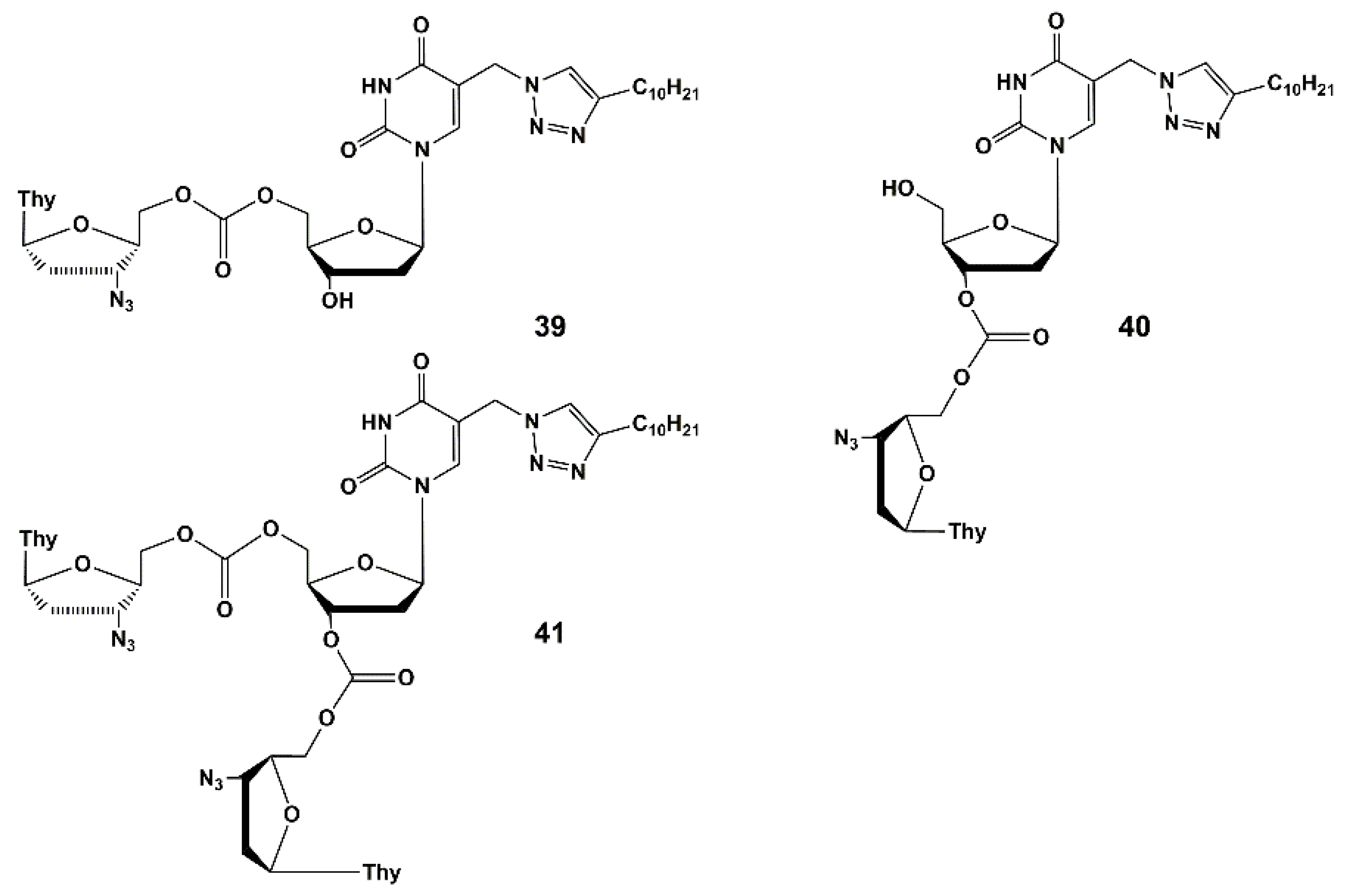
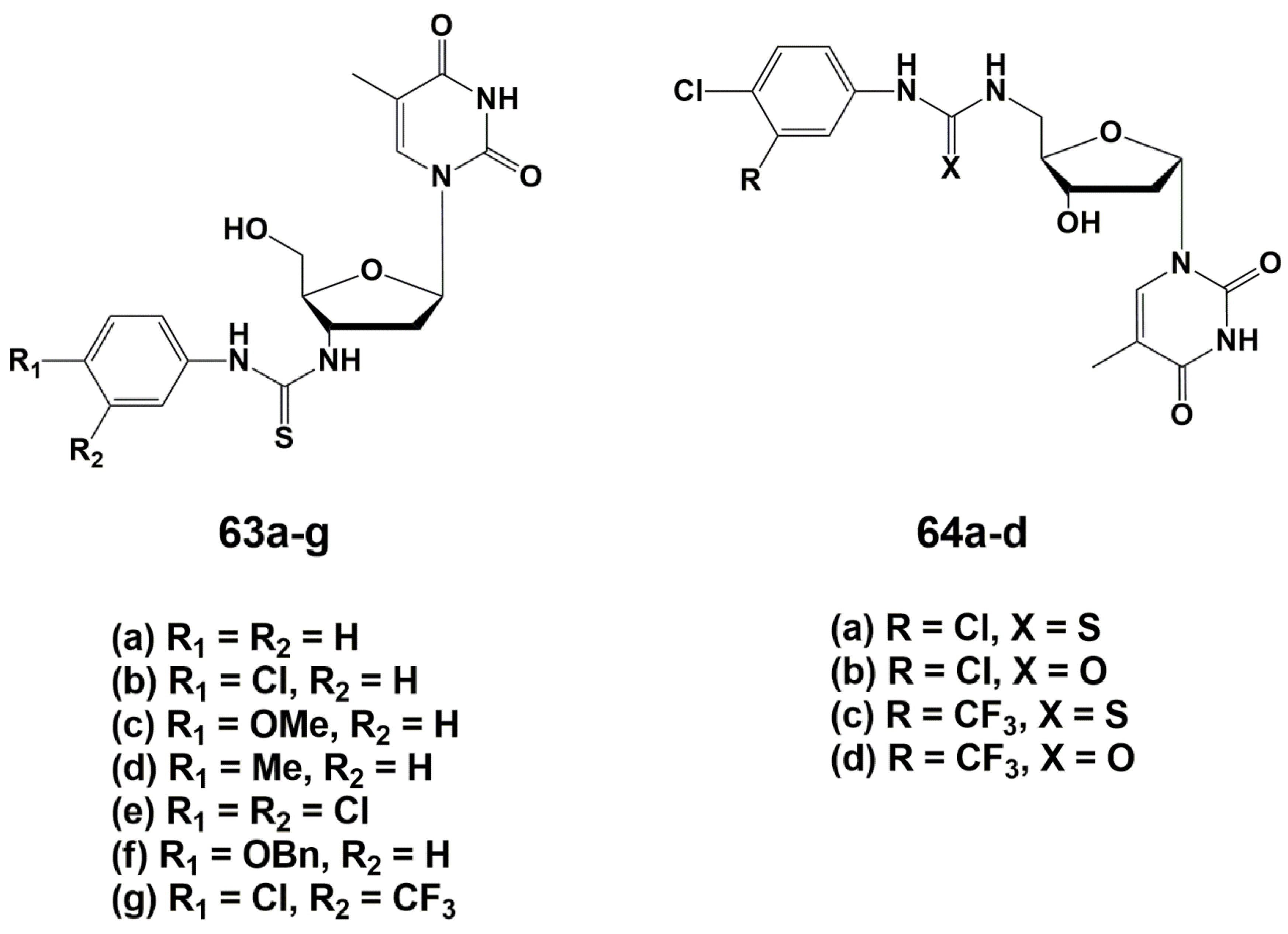
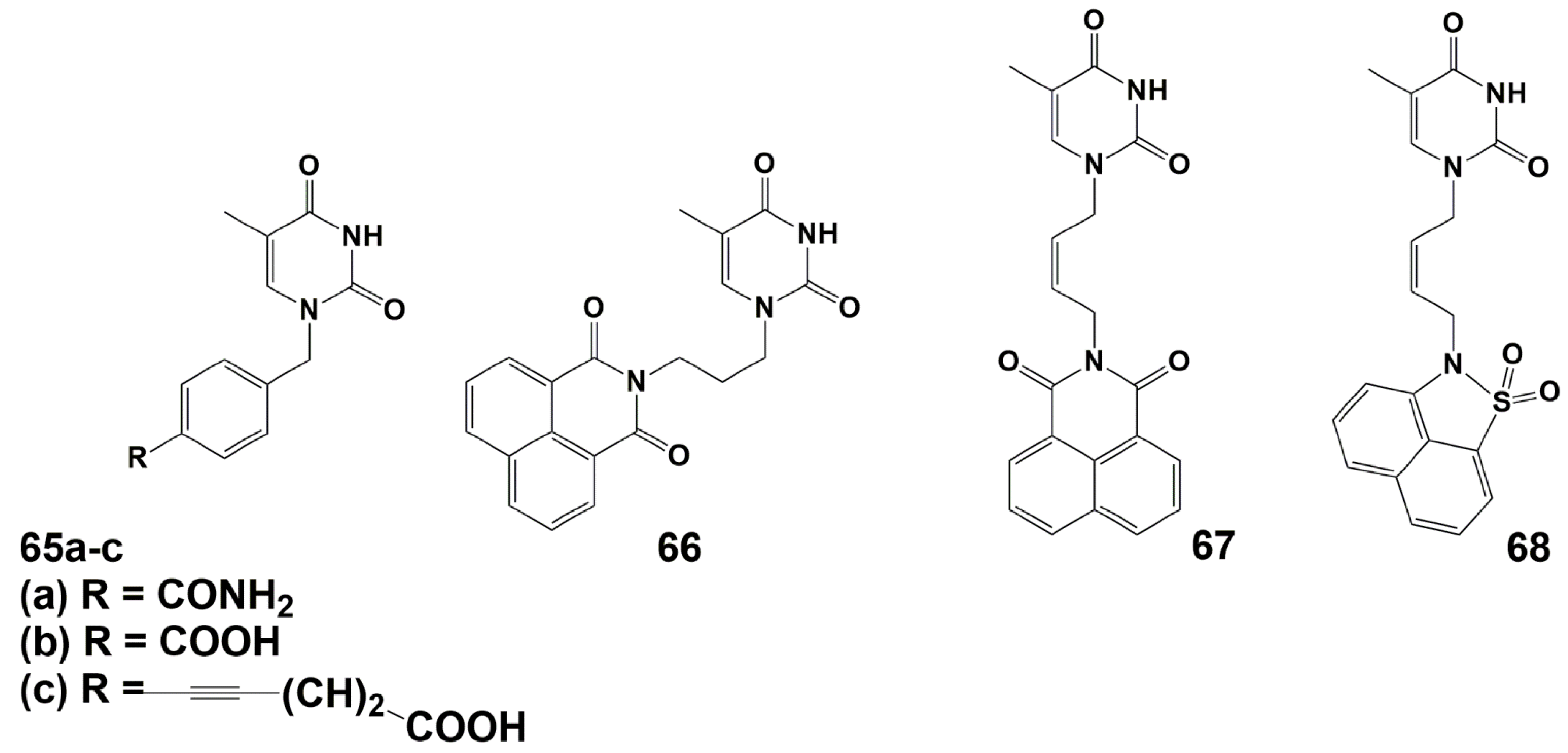
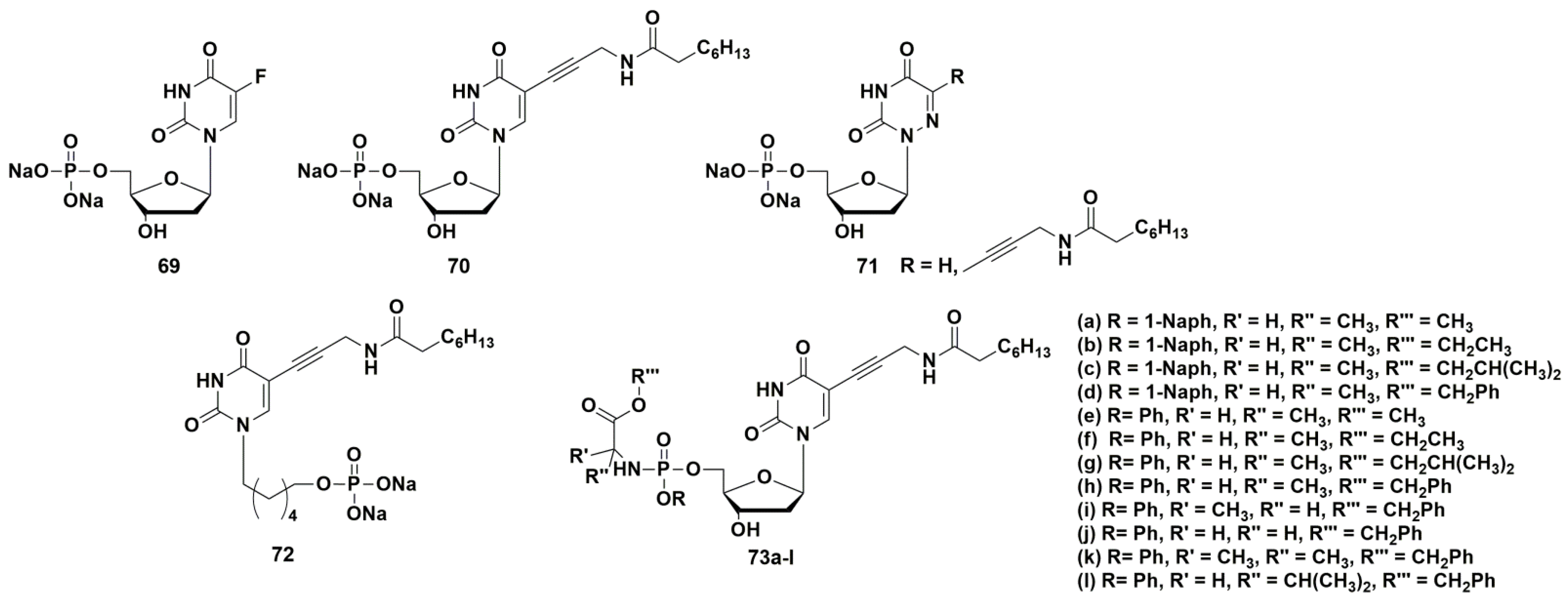
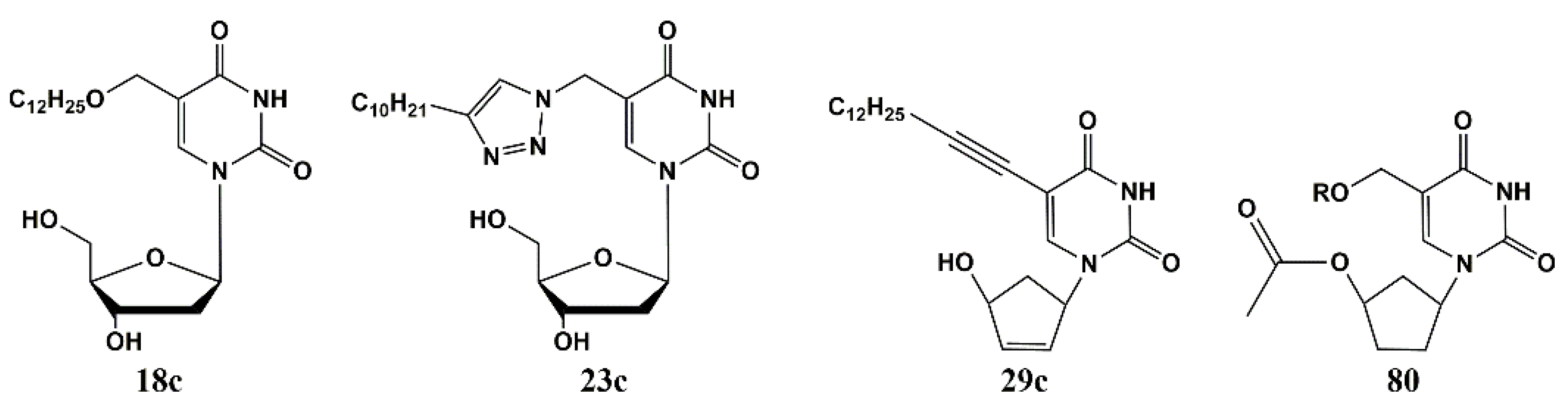
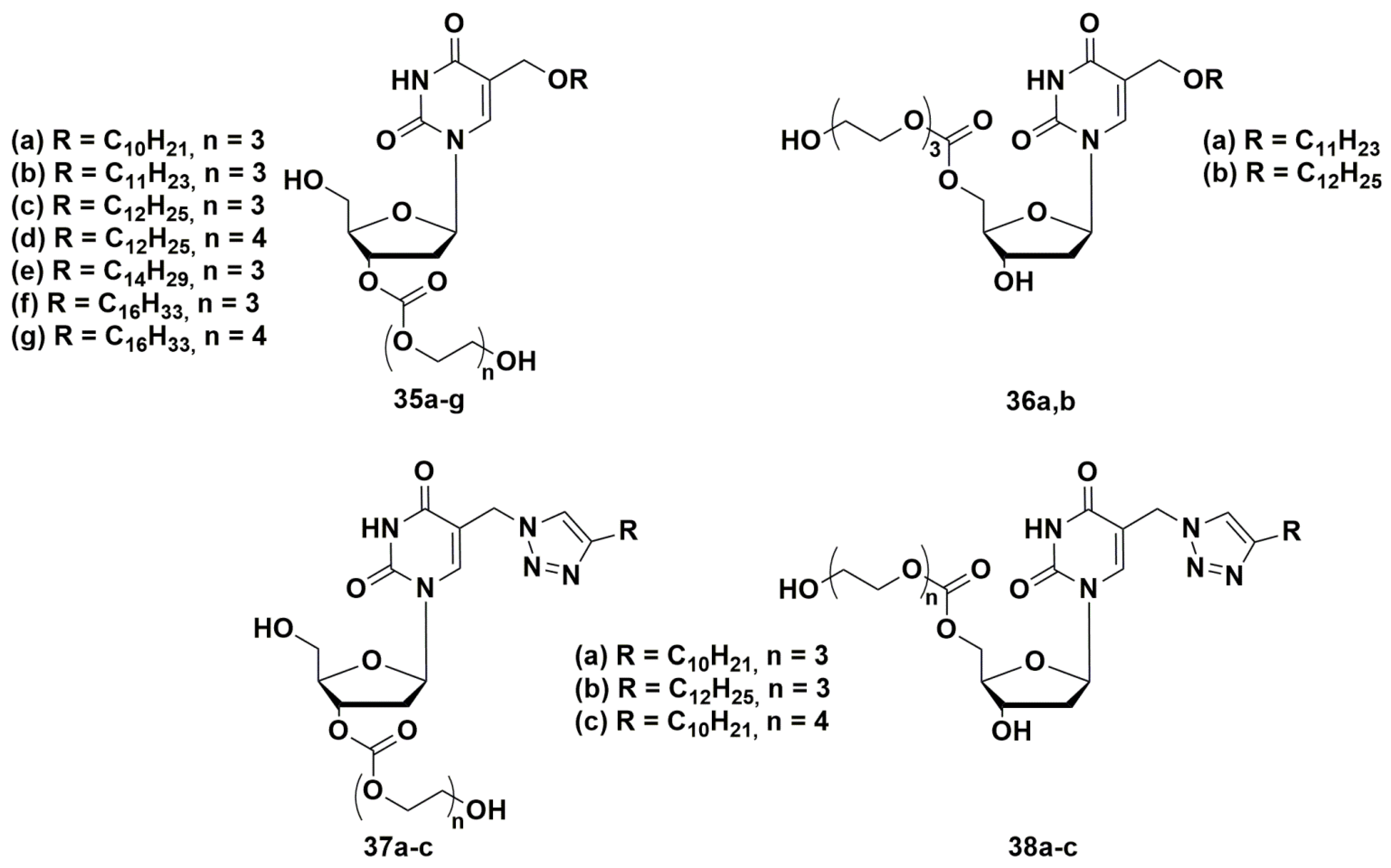
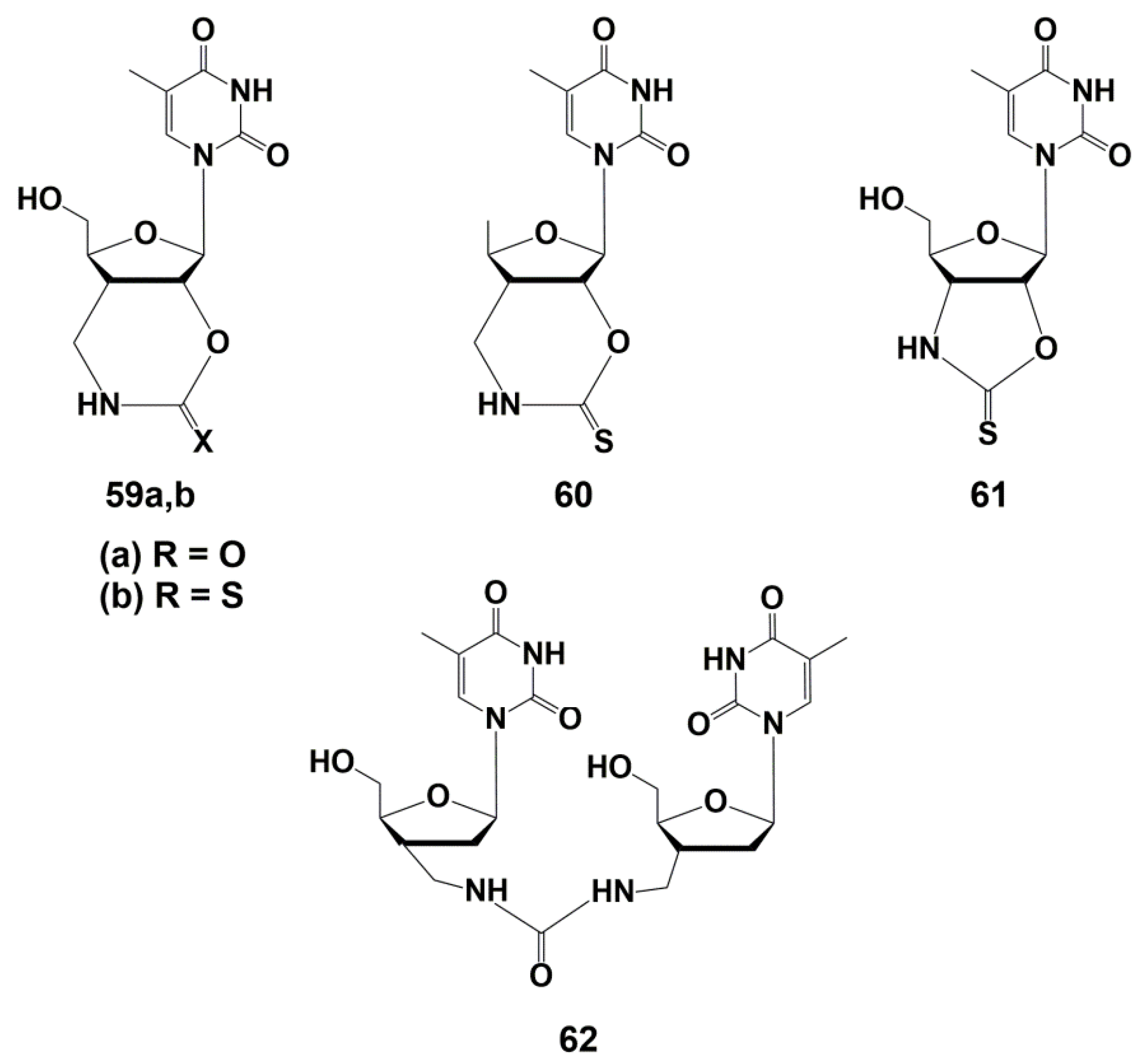
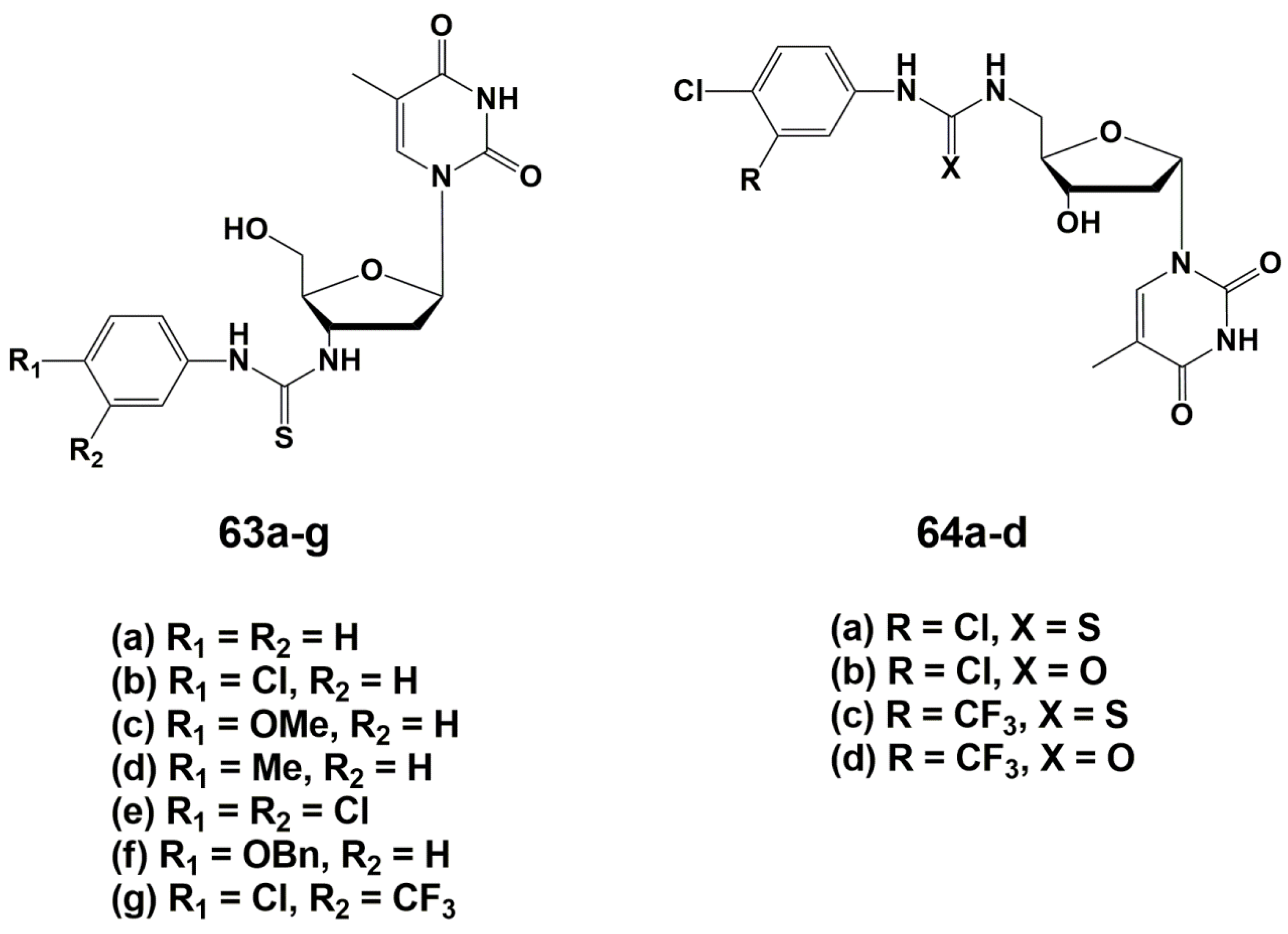
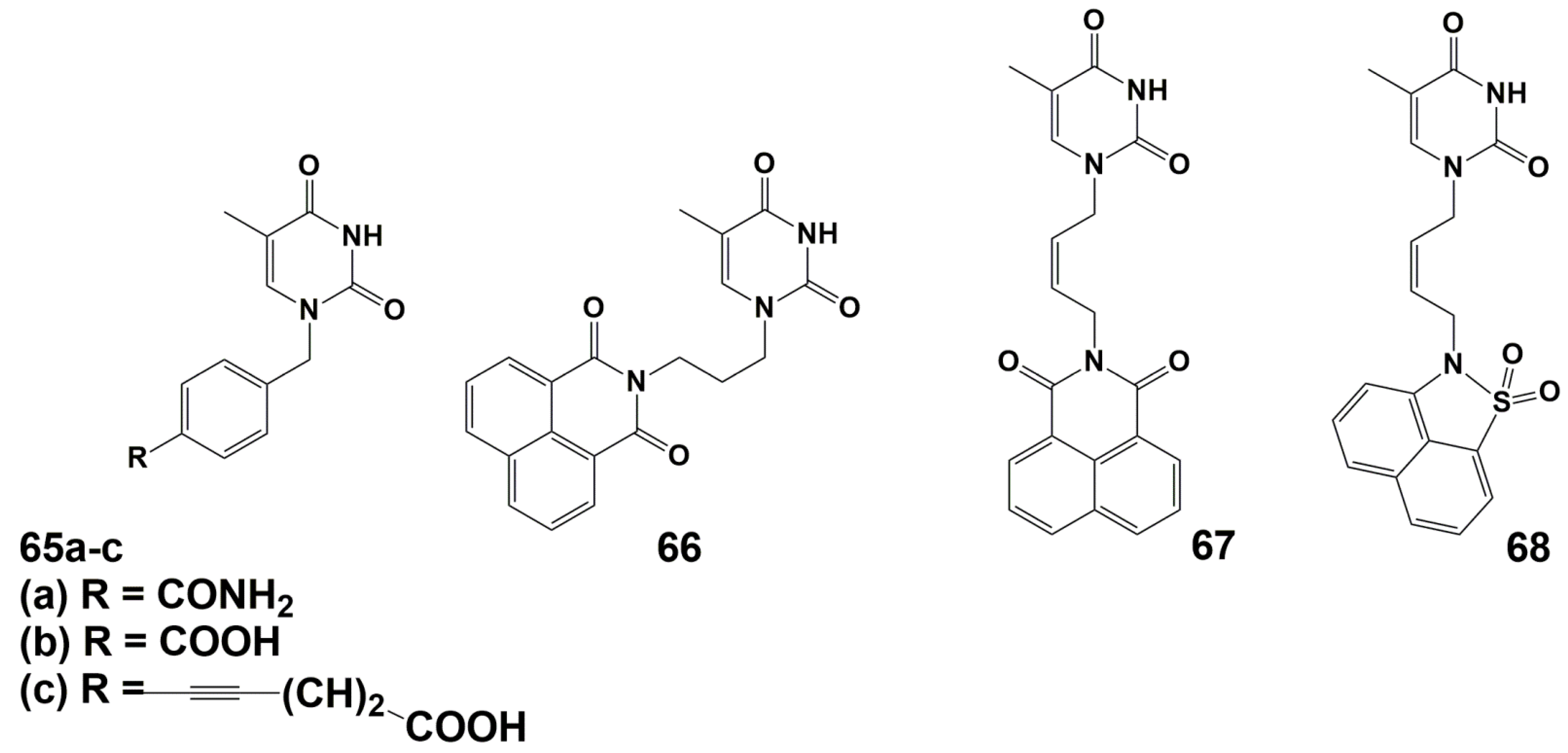
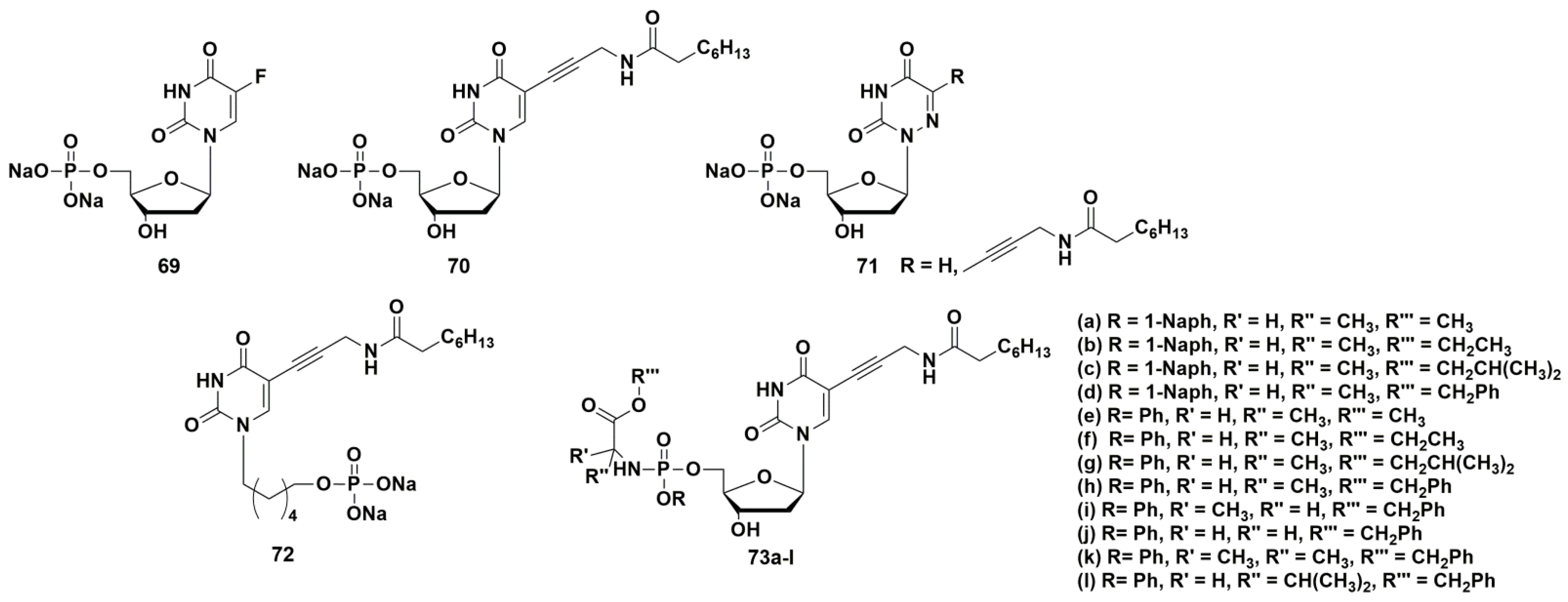
4.1. Depot Forms with Increased Solubility
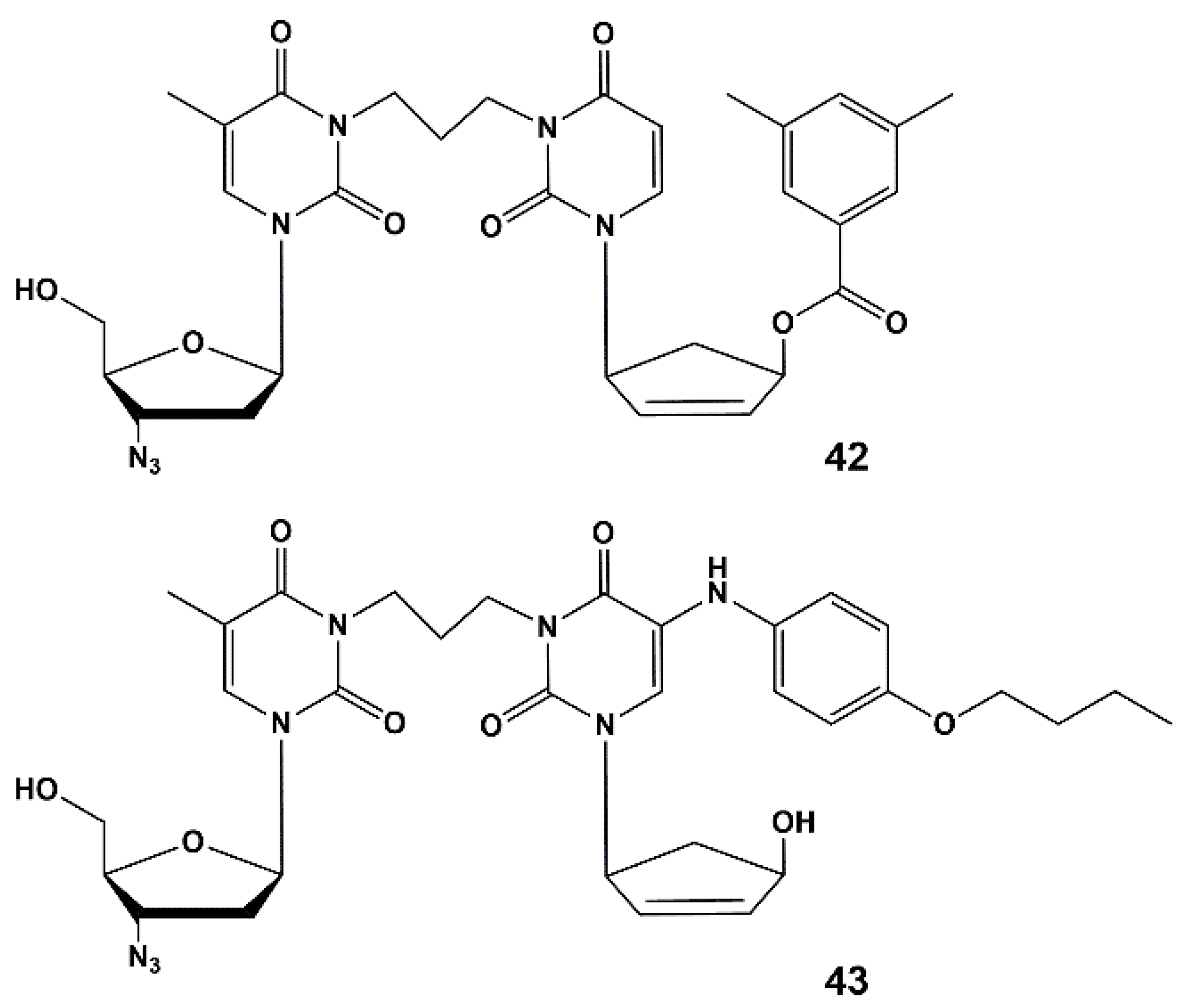
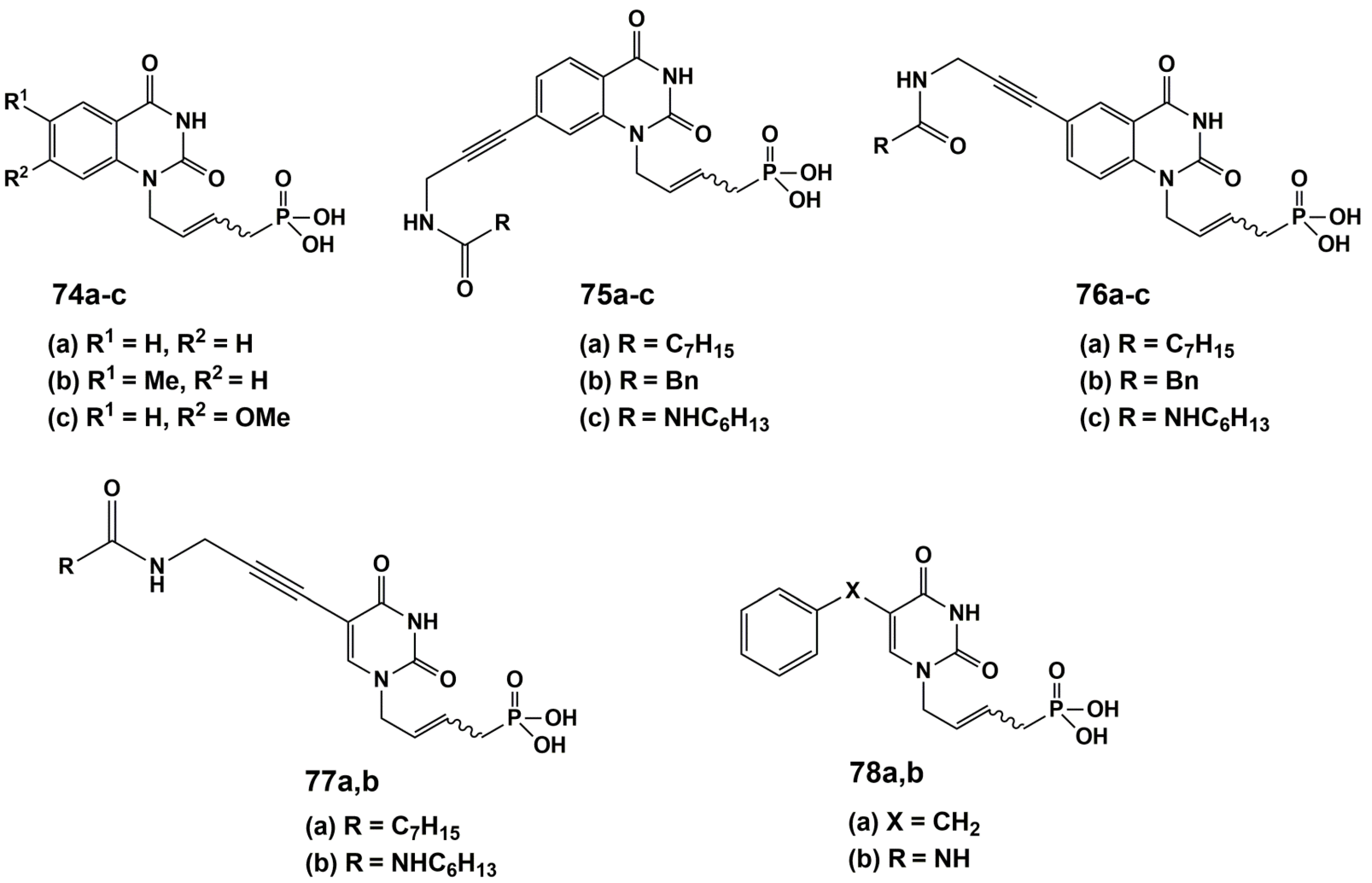
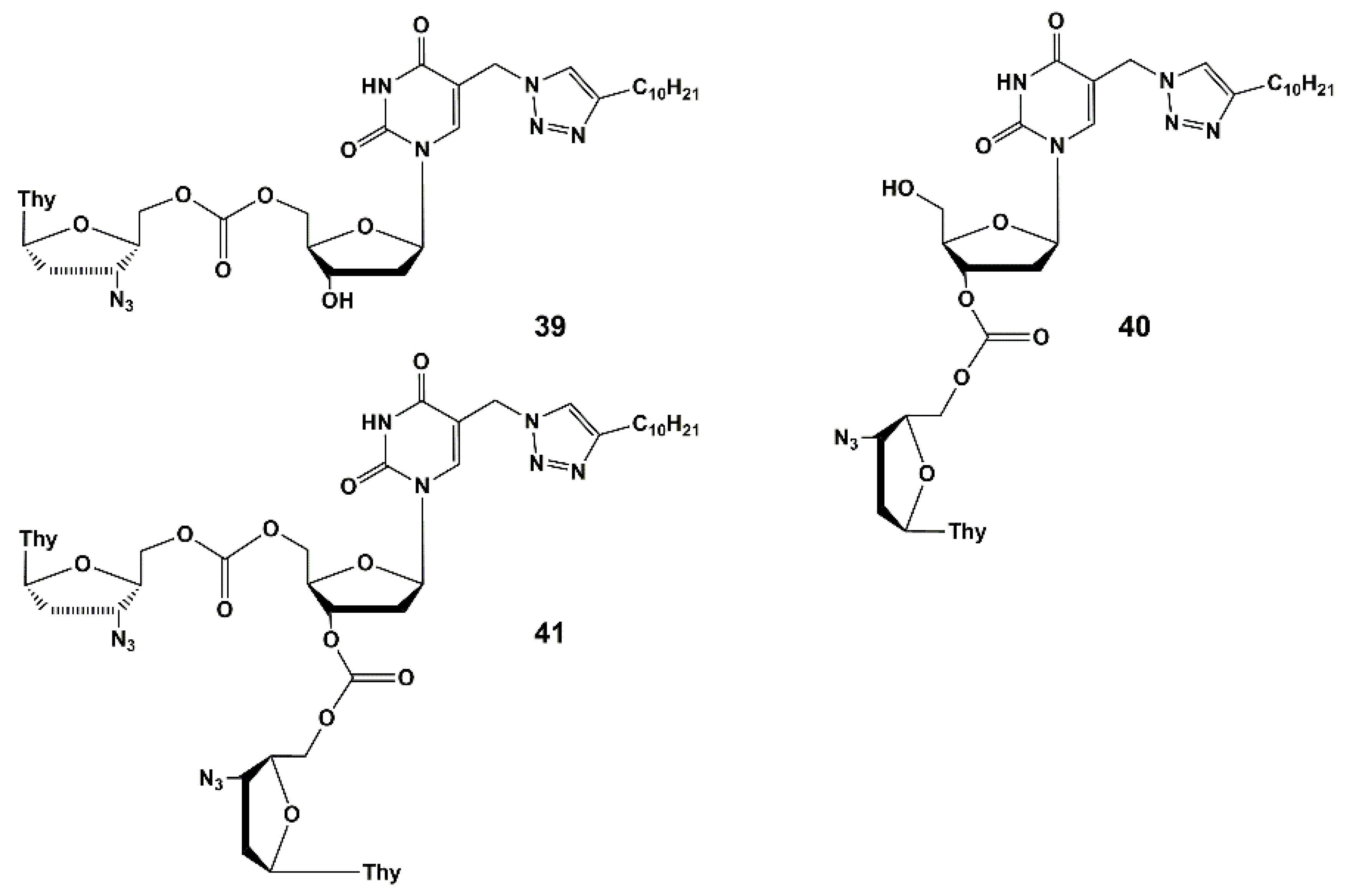
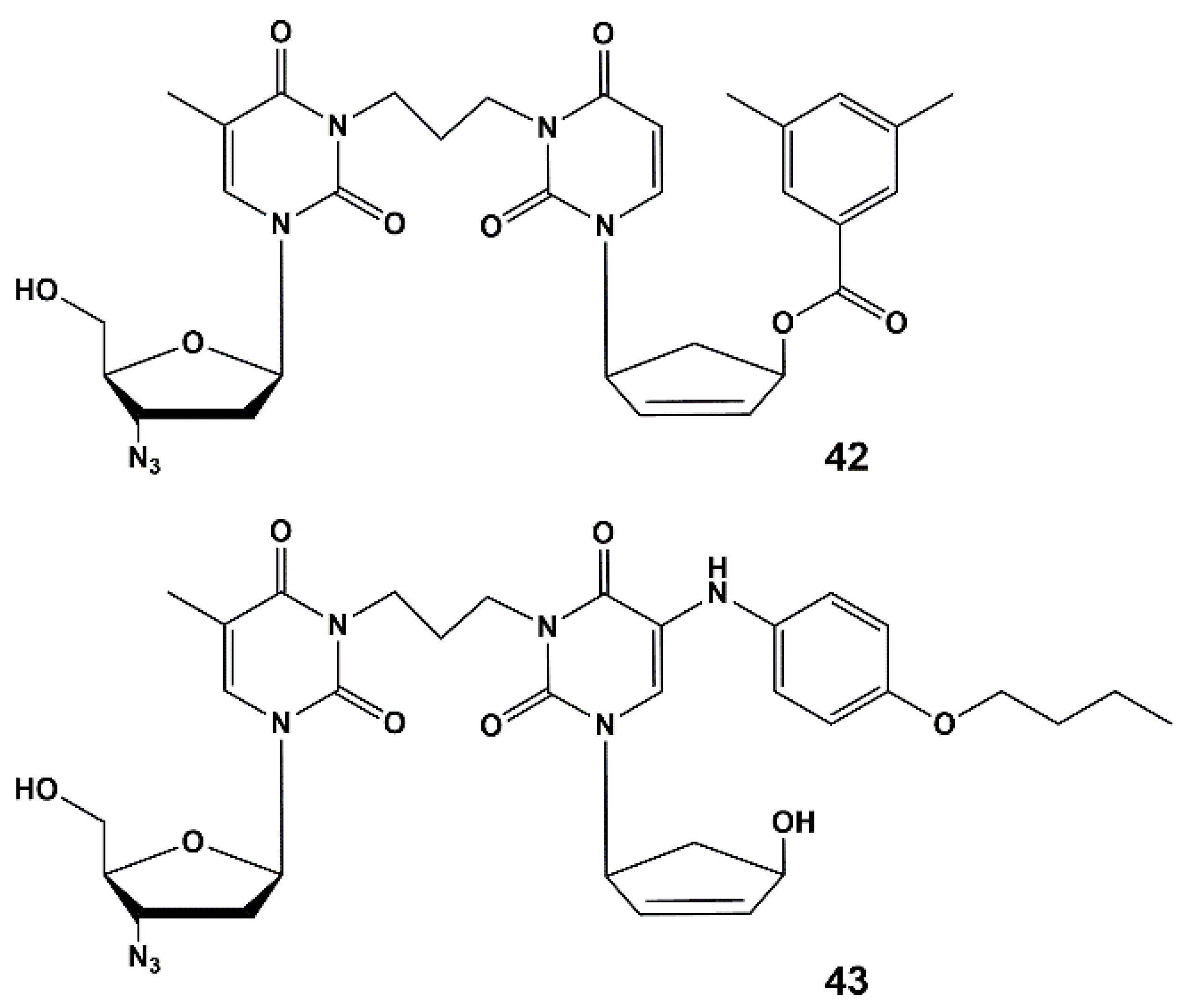
4.2. Dual Action Nucleoside Derivatives
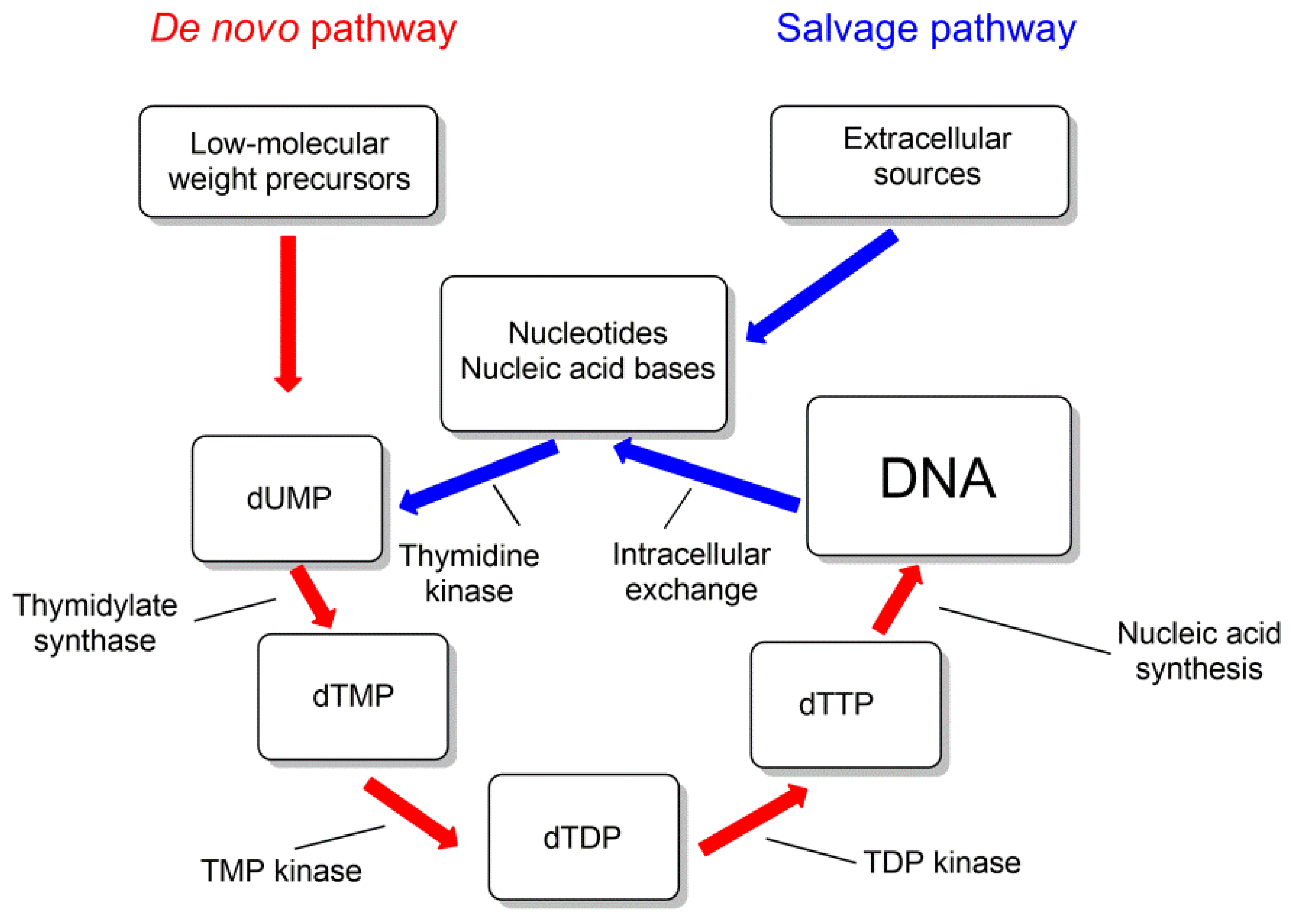
5. Target Enzymes for Nucleoside Inhibitors of M. tuberculosis
5.1. Thymidine Monophosphate Kinase Inhibitors
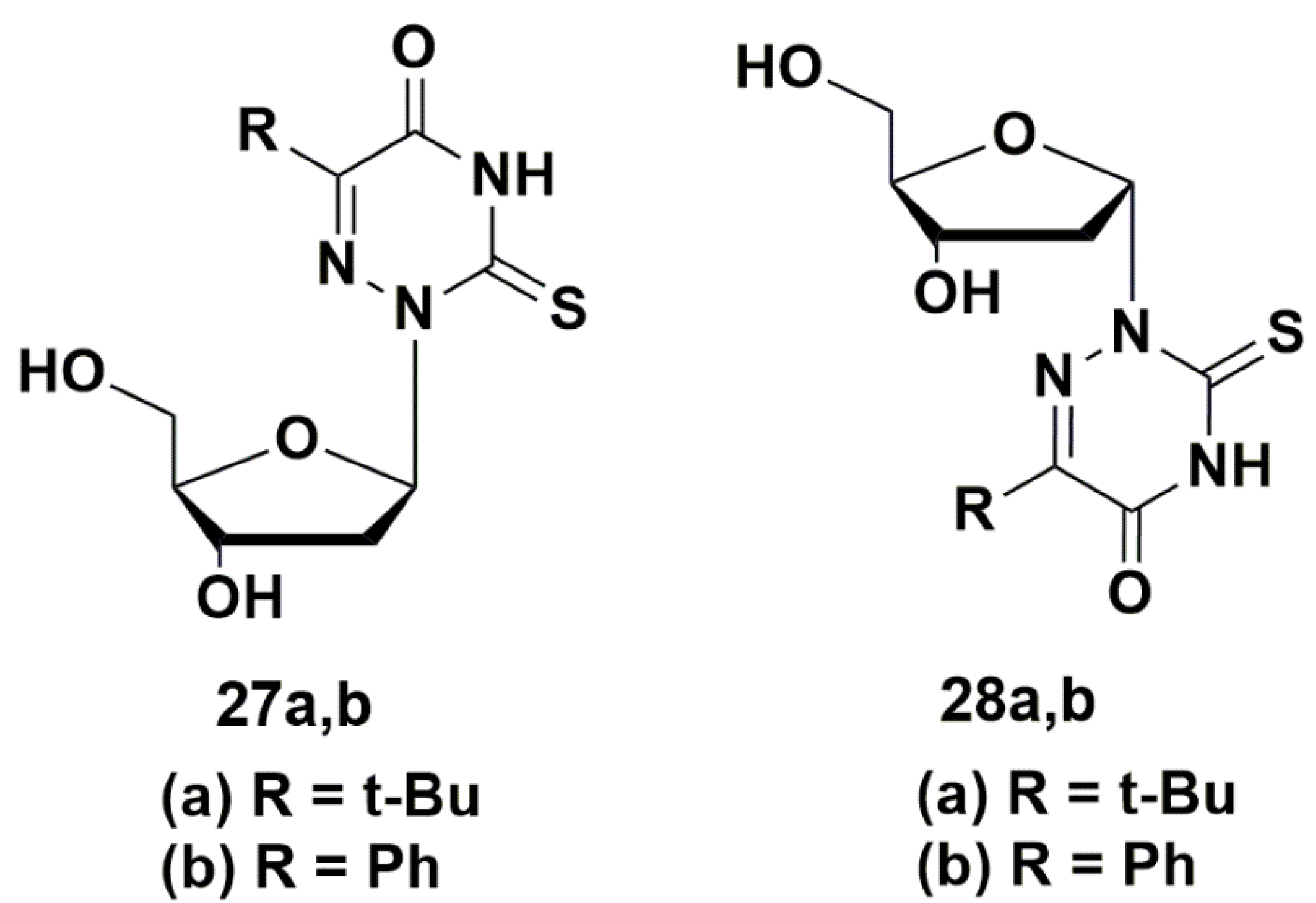
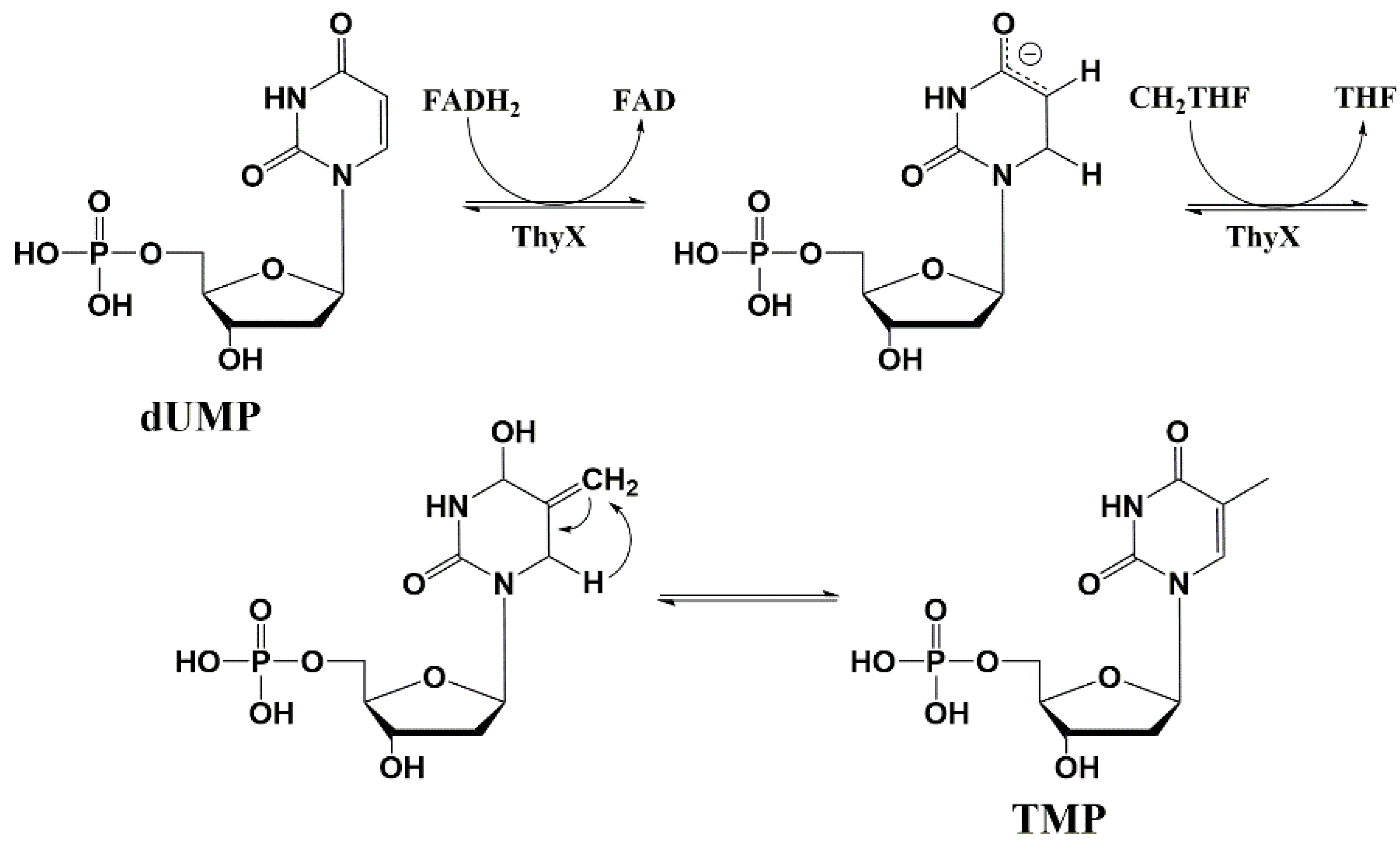
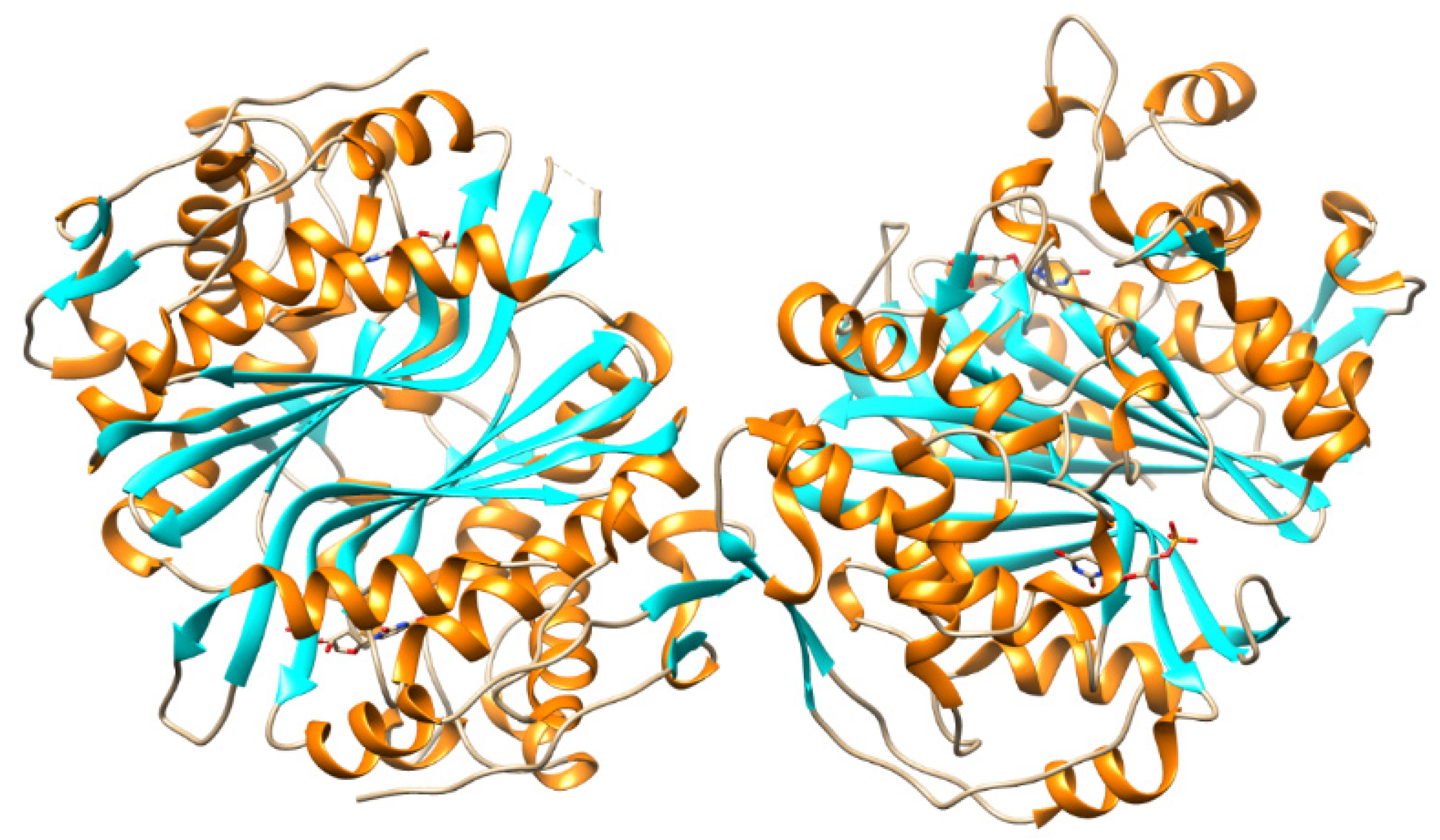
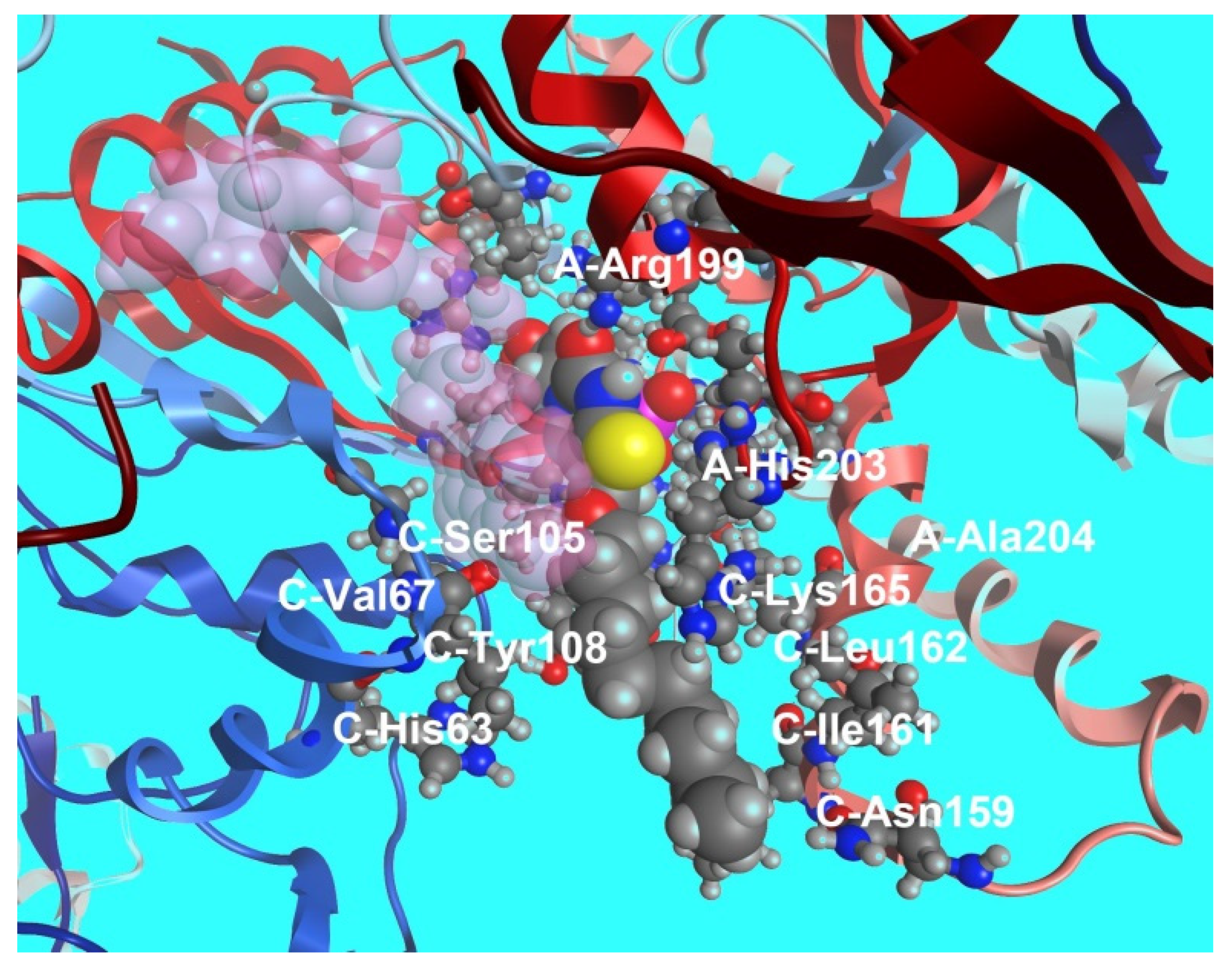
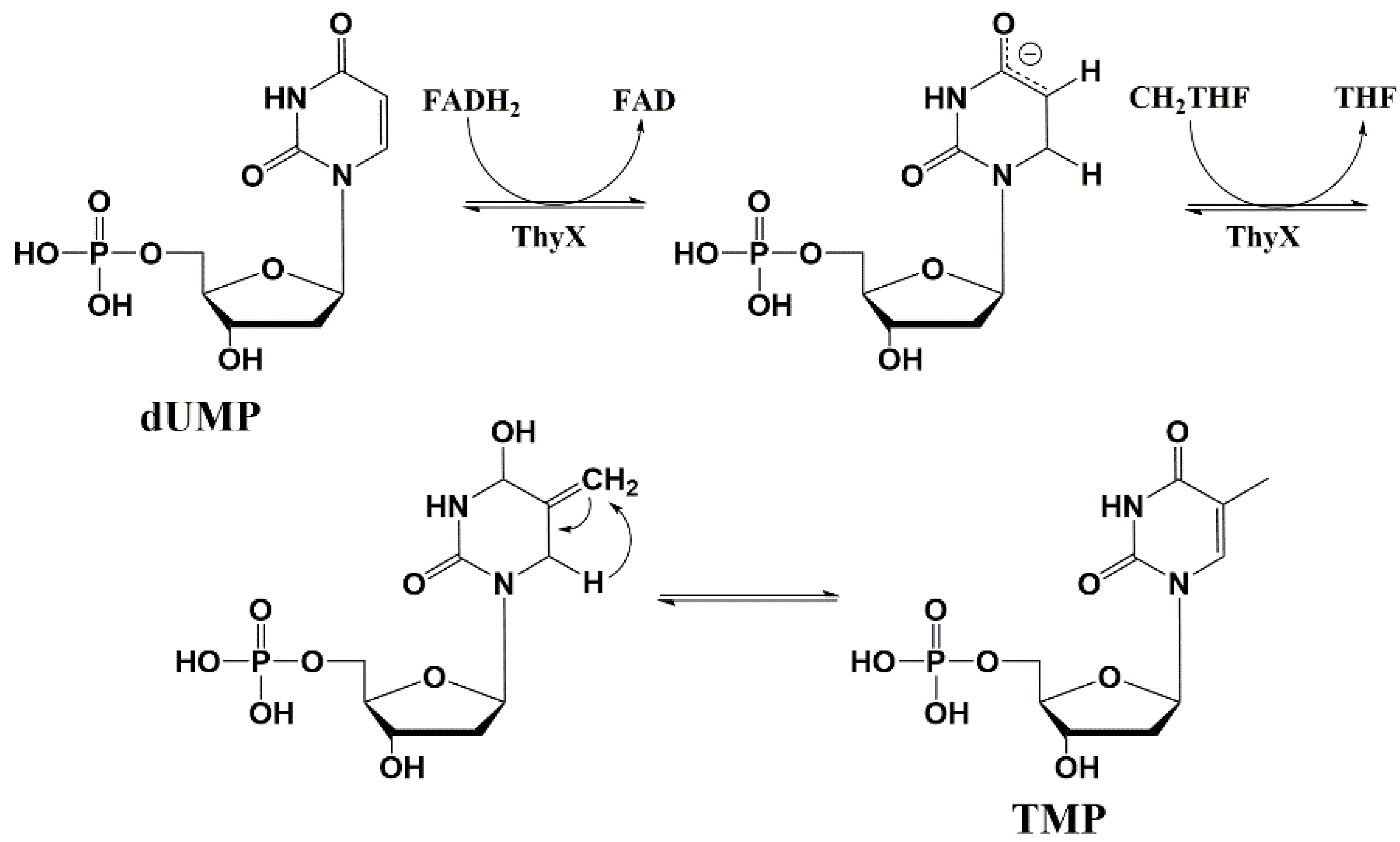
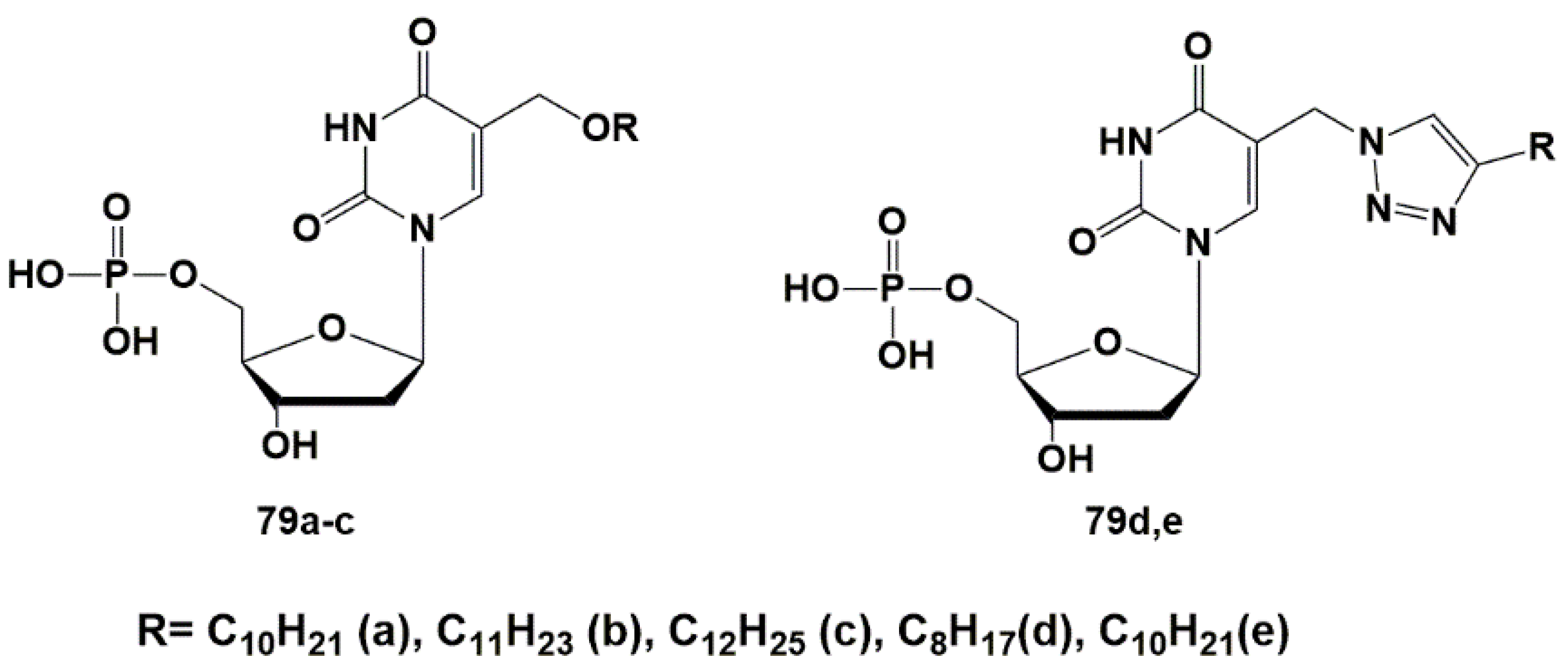
5.2. Nucleic Base Modification
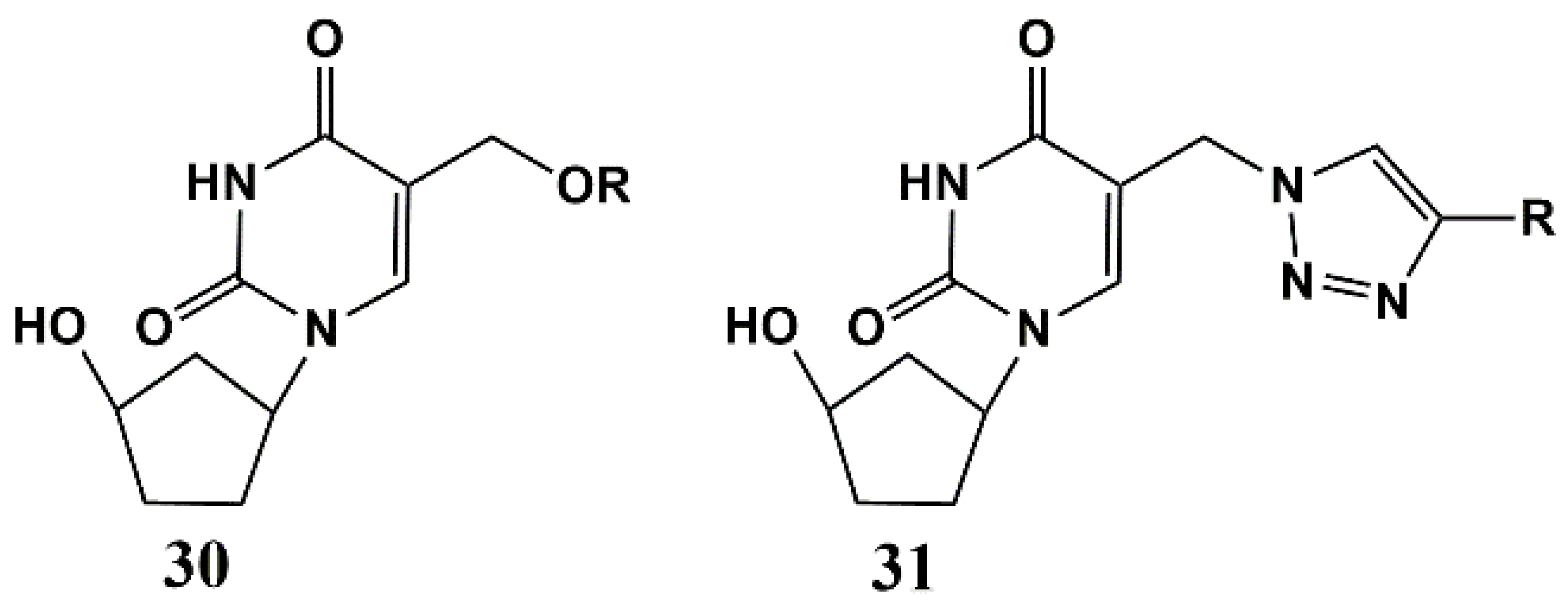
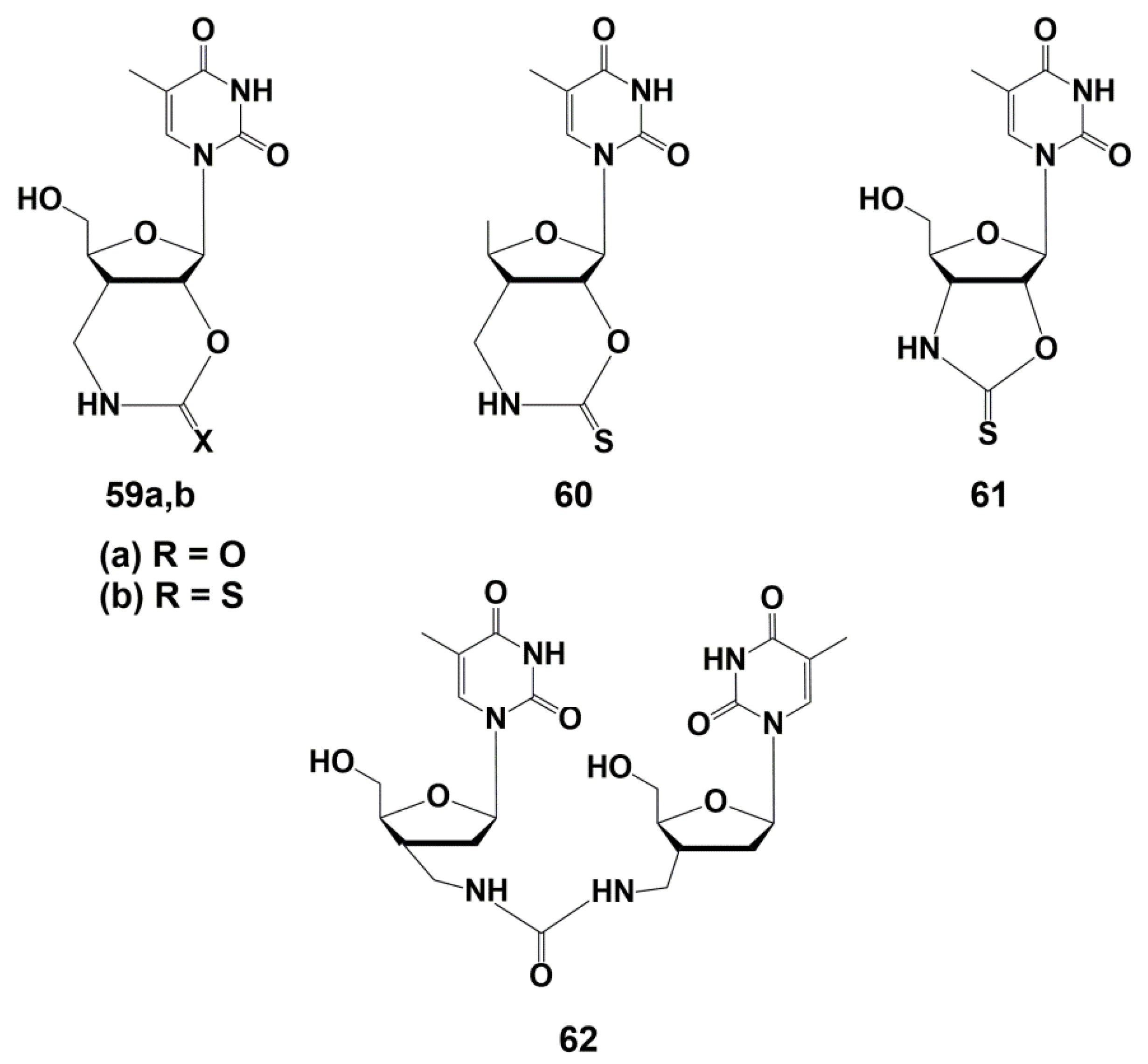
5.3. Modification of the Carbohydrate Fragment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
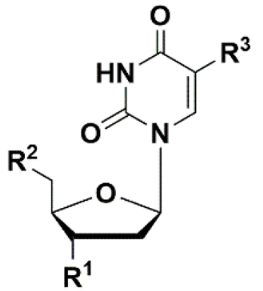 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ki (µM) TMPKmtb | Ref. |
| dTMP | OH | OPO32− | CH3 | 4.5 | [83,84] |
| Thymidine | OH | OH | CH3 | 27 | [83,84] |
| 44 | OH | OPO32− | Br | 30 * | [83,84] |
| 45 | OH | OPO32− | CH2OH | 110 | [85] |
| 46 | OH | OPO32− | Furan-2-yl | 140 | [85] |
| 47 | N3 | OPO32− | CH3 | 10 | [83,84,85,86] |
| 48 | NH2 | OPO32− | CH3 | 235 | [86] |
| 49 | CH2N3 | OPO32− | CH3 | 12 | [88] |
| 50 | CH2NH2 | OPO32− | CH3 | 10.5 | [88] |
| 51 | CH2F | OPO32− | CH3 | 15 | [88] |
| 52 | CH2OH | OPO32− | CH3 | 29 | [88] |
| 53 | OH | N3 | CH3 | 7 | [89] |
| 54 | OH | NH2 | CH3 | 11 | [89] |
| 55 | OH | OH | Br | 5 | [90] |
| 56 | OH | OH | Cl | 10 | [90] |
| 57 | N3 | OH | Br | 10.5 | [91] |
| 58 | N3 | OH | Cl | 16 | [91] |
5.4. Thymidylate Synthase Inhibitors
6. Potential Targets of the Antimicrobial Action for 5-Modified 2′-Deoxynucleoside Derivatives and Their 5′-Norcarbocyclic Analogues
7. Conclusions
- Two specific enzymes of Mtb were identified as targets for compounds with antimycobacterial activity: thymidine monophosphate kinase and thymidylate synthase Thy-X. A number of effective inhibitors were synthesized.
- Several series of nucleoside derivatives and analogues have been synthesized with high antimycobacterial activity, comparable to the activity of some anti-TB drugs currently used in clinic and effective against drug-resistant strains of Mtb. Some compounds can be considered as drug prototypes.
- Rational methods have been developed to increase the bioavailability of nucleoside derivatives and analogues with high antimycobacterial activity by design of their depot forms.
- An original approach was proposed for the synthesis of depot forms with dual-action, which was used for obtaining compounds simultaneously effective against HIV and Mtb.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient Origin and Gene Mosaicism of the Progenitor of Mycobacterium Tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report. Available online: www.who.int/health-topics/tuberculosis (accessed on 19 February 2022).
- WHO. Tuberculosis Factsheet, Online. Available online: https://www.who.int/publications/m/item/factsheet-global-tb-report-2021 (accessed on 21 February 2022).
- Martini, M.; Besozzi, G.; Barberis, I. The Never-Ending Story of the Fight against Tuberculosis: From Koch’s Bacillus to Global Control Programs. J. Prev. Med. Hyg. 2018, 59, E241–E247. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Sharma, D.; Singh, R. Tuberculosis and Its Treatment: An Overview. Mini-Rev. Med. Chem. 2017, 18, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dwivedi, S.P.; Gaharwar, U.S.; Meena, R.; Rajamani, P.; Prasad, T. Recent Updates on Drug Resistance in Mycobacterium Tuberculosis. J. Appl. Microbiol. 2020, 128, 1547–1567. [Google Scholar] [CrossRef] [Green Version]
- Makarov, V.; Salina, E.; Reynolds, R.C.; Kyaw Zin, P.P.; Ekins, S. Molecule Property Analyses of Active Compounds for Mycobacterium Tuberculosis. J. Med. Chem. 2020, 63, 8917–8955. [Google Scholar] [CrossRef]
- Hoffman, P.S. Antibacterial Discovery: 21st Century Challenges. Antibiotics 2020, 9, 213. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Cohen, A.; Mathiasen, V.D.; Schön, T.; Wejse, C. The Global Prevalence of Latent Tuberculosis: A Systematic Review and Meta-Analysis. Eur. Respir. J. 2019, 54, 1900655. [Google Scholar] [CrossRef]
- Datta, S.; Evans, C.A. Healthy Survival after Tuberculosis. Lancet Infect. Dis. 2019, 19, 1045–1047. [Google Scholar] [CrossRef]
- Martinez, L.; Verma, R.; Croda, J.; Horsburgh, C.R.; Walter, K.S.; Degner, N.; Middelkoop, K.; Koch, A.; Hermans, S.; Warner, D.F.; et al. Detection, Survival and Infectious Potential of Mycobacterium Tuberculosis in the Environment: A Review of the Evidence and Epidemiological Implications. Eur. Respir. J. 2019, 53, 1802302. [Google Scholar] [CrossRef] [PubMed]
- Joean, O.; Thiele, T.; Schütz, K.; Schwerk, N.; Sedlacek, L.; Kalsdorf, B.; Baumann, U.; Stoll, M. Multidrug-Resistant Mycobacterium Tuberculosis: A Report of Cosmopolitan Microbial Migration and an Analysis of Best Management Practices. BMC Infect. Dis. 2020, 20, 678. [Google Scholar] [CrossRef] [PubMed]
- Heyckendorf, J.; Lange, C.; Martensen, J. Multidrug-Resistant Tuberculosis. In Emerging Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2014; pp. 239–253. [Google Scholar] [CrossRef]
- Khan, M.T.; Junaid, M.; Mao, X.; Wang, Y.; Hussain, A.; Malik, S.I.; Wei, D. Pyrazinamide Resistance and Mutations L19R, R140H, and E144K in Pyrazinamidase of Mycobacterium Tuberculosis. J. Cell. Biochem. 2019, 120, 7154–7166. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Khan, A.; Ali, S.S.; Ali, S.; Wei, D.-Q. A Computational Perspective on the Dynamic Behaviour of Recurrent Drug Resistance Mutations in the PncA Gene from Mycobacterium Tuberculosis. RSC Adv. 2021, 11, 2476–2486. [Google Scholar] [CrossRef]
- Alanis, A.J. Resistance to Antibiotics: Are We in the Post-Antibiotic Era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef]
- Yssel, A.E.J.; Vanderleyden, J.; Steenackers, H.P. Repurposing of Nucleoside- and Nucleobase-Derivative Drugs as Antibiotics and Biofilm Inhibitors. J. Antimicrob. Chemother. 2017, 72, 2156–2170. [Google Scholar] [CrossRef]
- Winn, M.; Goss, R.J.M.; Kimura, K.; Bugg, T.D.H. Antimicrobial Nucleoside Antibiotics Targeting Cell Wall Assembly: Recent Advances in Structure–Function Studies and Nucleoside Biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [Google Scholar] [CrossRef]
- Duckworth, B.P.; Nelson, K.M.; Aldrich, C.C. Adenylating Enzymes in Mycobacterium Tuberculosis as Drug Targets. Curr. Top. Med. Chem. 2012, 12, 766–796. [Google Scholar] [CrossRef]
- Duckworth, B.P.; Wilson, D.J.; Nelson, K.M.; Boshoff, H.I.; Barry, C.E.; Aldrich, C.C. Development of a Selective Activity-Based Probe for Adenylating Enzymes: Profiling MbtA Involved in Siderophore Biosynthesis from Mycobacterium Tuberculosis. ACS Chem. Biol. 2012, 7, 1653–1658. [Google Scholar] [CrossRef] [Green Version]
- Shakya, N.; Garg, G.; Agrawal, B.; Kumar, R. Chemotherapeutic Interventions Against Tuberculosis. Pharmaceuticals 2012, 5, 690–718. [Google Scholar] [CrossRef] [Green Version]
- Shmalenyuk, E.R.; Kochetkov, S.N.; Alexandrova, L.A. Novel Inhibitors of Mycobacterium tuberculosis Growth Based on Modified Pyrimidine Nucleosides and Their Analogues. Russ. Chem. Rev. 2013, 82, 896–915. [Google Scholar] [CrossRef]
- Van Calenbergh, S.; Pochet, S.; Munier-Lehmann, H. Drug Design and Identification of Potent Leads Against Mycobacterium tuberculosis Thymidine Monophosphate Kinase. Curr. Top. Med. Chem. 2012, 12, 694–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside Derived Antibiotics to Fight Microbial Drug Resistance: New Utilities for an Established Class of Drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Serpi, M. Nucleoside Analogs and Tuberculosis: New Weapons against an Old Enemy. Future Med. Chem. 2015, 7, 291–314. [Google Scholar] [CrossRef]
- Ducati, R.G.; Breda, A.; Basso, L.A.; Santos, D.S. Purine Salvage Pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1258–1275. [Google Scholar] [CrossRef]
- Villela, A.D.; Sanchez-Quitian, Z.A.; Ducati, R.G.; Santos, D.S.; Basso, L.A. Pyrimidine Salvage Pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 2011, 18, 1286–1298. [Google Scholar] [CrossRef]
- Dawadi, S.; Kawamura, S.; Rubenstein, A.; Remmel, R.; Aldrich, C.C. Synthesis and Pharmacological Evaluation of Nucleoside Prodrugs Designed to Target Siderophore Biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Dawadi, S.; Boshoff, H.I.M.; Park, S.W.; Schnappinger, D.; Aldrich, C.C. Conformationally Constrained Cinnolinone Nucleoside Analogues as Siderophore Biosynthesis Inhibitors for Tuberculosis. ACS Med. Chem. Lett. 2018, 9, 386–391. [Google Scholar] [CrossRef]
- Johar, M.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Synthesis and in Vitro Anti-Mycobacterial Activity of 5-Substituted Pyrimidine Nucleosides. Bioorg. Med. Chem. 2005, 13, 6663–6671. [Google Scholar] [CrossRef]
- Kumar, R.; Kunimoto, D.; Manning, T.; Srivastav, N. In Vitro Anti-Mycobacterial Activities of Various 2′-Deoxyuridine, 2′-Arabinouridine and 2′-Arabinofluoro-2′-Deoxyuridine Analogues: Synthesis and Biological Studies. Med. Chem. 2006, 2, 287–293. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Design and Studies of Novel 5-Substituted Alkynylpyrimidine Nucleosides as Potent Inhibitors of Mycobacteria. J. Med. Chem. 2005, 48, 7012–7017. [Google Scholar] [CrossRef] [PubMed]
- Johar, M.; Manning, T.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Growth Inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium In Vitro: Effect of 1-β-d-2′-Arabinofuranosyl and 1-(2′-Deoxy-2′-Fluoro-β-d-2′-Ribofuranosyl) Pyrimidine Nucleoside Analogs. J. Med. Chem. 2007, 50, 3696–3705. [Google Scholar] [CrossRef]
- Rai, D.; Johar, M.; Srivastav, N.C.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Inhibition of Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium avium by Novel Dideoxy Nucleosides. J. Med. Chem. 2007, 50, 4766–4774. [Google Scholar] [CrossRef] [PubMed]
- Srivastav, N.C.; Manning, T.; Kunimoto, D.Y.; Kumar, R. Studies on Acyclic Pyrimidines as Inhibitors of Mycobacteria. Bioorg. Med. Chem. 2007, 15, 2045–2053. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Sonogashira, K. Development of Pd–Cu Catalyzed Cross-Coupling of Terminal Acetylenes with Sp2-Carbon Halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Shakya, N.; Srivastav, N.C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. 3′-Bromo Analogues of Pyrimidine Nucleosides as a New Class of Potent Inhibitors of Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Shakya, N.; Srivastav, N.C.; Bhavanam, S.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Discovery of Novel 5-(Ethyl or Hydroxymethyl) Analogs of 2′-‘up’ Fluoro (or Hydroxyl) Pyrimidine Nucleosides as a New Class of Mycobacterium tuberculosis, Mycobacterium bovis and Mycobacterium avium Inhibitors. Bioorg. Med. Chem. 2012, 20, 4088–4097. [Google Scholar] [CrossRef]
- Platonova, Y.B.; Volov, A.N.; Tomilova, L.G. The Synthesis and Antituberculosis Activity of 5-Alkynyl Uracil Derivatives. Bioorg. Med. Chem. Lett. 2020, 30, 127351. [Google Scholar] [CrossRef]
- Volov, A.N.; Volov, N.A.; Platonova, Y.B. Design and Synthesis of Novel 5-Alkynyl Pyrimidine Nucleosides Derivatives: Influence of C-6-Substituent on Antituberculosis Activity. Bioorg. Med. Chem. Lett. 2021, 48, 128261. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Shmalenyuk, E.R.; Kochetkov, S.N.; Erokhin, V.V.; Smirnova, T.G.; Andreevskaia, S.N.; Chernousova, L.N. New 5-Modified Pyrimidine Nucleoside Inhibitors of Mycobacterial Growth. Acta Naturae 2010, 2, 108–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmalenyuk, E.R.; Chernousova, L.N.; Karpenko, I.L.; Kochetkov, S.N.; Smirnova, T.G.; Andreevskaya, S.N.; Chizhov, A.O.; Efremenkova, O.V.; Alexandrova, L.A. Inhibition of Mycobacterium tuberculosis Strains H37Rv and MDR MS-115 by a New Set of C5 Modified Pyrimidine Nucleosides. Bioorg. Med. Chem. 2013, 21, 4874–4884. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Efremenkova, O.V.; Andronova, V.L.; Galegov, G.A.; Solyev, P.N.; Karpenko, I.L.; Kochetkov, S.N. 5-(4-Alkyl-1,2,3-Triazol-1-Yl)Methyl Derivatives of 2′-Deoxyuridine as Inhibitors of Viral and Bacterial Growth. Russ. J. Bioorg. Chem. 2016, 42, 677–684. [Google Scholar] [CrossRef]
- Shmalenyuk, E.R.; Karpenko, I.L.; Chernousova, L.N.; Chizhov, A.O.; Smirnova, T.G.; Andreevskaya, S.N.; Alexandrova, L.A. New 5-Modified 2′-Deoxyuridine Derivatives: Synthesis and Antituberculosis Activity. Russ. Chem. Bull. 2014, 63, 1197–1200. [Google Scholar] [CrossRef]
- Negrya, S.D.; Efremenkova, O.V.; Solyev, P.N.; Chekhov, V.O.; Ivanov, M.A.; Sumarukova, I.G.; Karpenko, I.L.; Kochetkov, S.N.; Alexandrova, L.A. Novel 5-Substituted Derivatives of 2′-Deoxy-6-Azauridine with Antibacterial Activity. J. Antibiot. 2019, 72, 535–544. [Google Scholar] [CrossRef]
- Matyugina, E.; Khandazhinskaya, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Chizhov, A.; Karpenko, I.; Kochetkov, S.; Alexandrova, L. The Synthesis and Antituberculosis Activity of 5′-nor Carbocyclic Uracil Derivatives. Bioorg. Med. Chem. 2012, 20, 6680–6686. [Google Scholar] [CrossRef]
- Kusaka, T.; Yamamoto, H.; Shibata, M.; Muroi, M.; Kishi, T.; Mizuno, K. Streptomyces citricolor nov. Sp. and a new antibiotic, aristeromycin. J. Antibiot. 1968, 21, 255–263. [Google Scholar] [CrossRef]
- Hayashi, M.; Yaginuma, S.; Yoshioka, H.; Nakatsu, K. Studies on Neplanocin A, New Antitumor Antibiotic. II. Structure Determination. J. Antibiot. 1981, 34, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.I.; Caamaño, O.; Fernández, F.; Gómez, M.; Balzarini, J.; De Clercq, E. Synthesis, antiviral and cytostatic activities, of carbocyclic nucleosides incorporating a modified cyclopentane ring. IV. Adenosine and uridine analogues. Nucleosides Nucleotides Nucleic Acids 2002, 21, 243–255. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N. Carbocyclic Nucleoside Analogues: Classification, Target Enzymes, Mechanisms of Action and Synthesis. Russ. Chem. Rev. 2012, 81, 729–746. [Google Scholar] [CrossRef]
- Novikov, M.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Paramonova, M.P.; Ivanov, A.V.; Gavryushov, S.A.; Khandazhinskaya, A.L.; Kochetkov, S.N.; Pannecouque, C.; Andrei, G.; et al. N1,N3-Disubstituted Uracils as Nonnucleoside Inhibitors of HIV-1 Reverse Transcriptase. Bioorg. Med. Chem. 2013, 21, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T. S-Adenosyl-L-Methionine-Dependent Macromolecule Methyltransferases: Potential Targets for the Design of Chemotherapeutic Agents. J. Med. Chem. 1980, 23, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T.; Creveling, C.R.; Ueland, P.M. (Eds.) Biological Methylation and Drug Design; Humana Press: Totowa, NJ, USA, 1986; ISBN 978-1-4612-9398-9. [Google Scholar]
- Yuan, C.-S.; Liu, S.; Wnuk, S.F.; Robins, M.J.; Borchardt, R.T. Design and Synthesis of S-Adenosylhomocysteine Hydrolase Inhibitors as Broad-Spectrum Antiviral Agents. In Advances in Antiviral Drug Design; De Clercq, E., Ed.; Elsevier: Paris, France, 1996; pp. 41–88. ISBN 1-55938-693-2. [Google Scholar]
- Schneller, S.W.; Seley, K.L.; Hegde, V.R.; Rajappan, V.P. 5′-Norcarbanucleosides in L-Like Configurations. In Recent Advances in Nucleosides: Chemistry and Chemotherapy; Chu, C.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2002; pp. 291–297. ISBN 0-444-50951-8. [Google Scholar]
- Hegde, V.R.; Seley, K.L.; Schneller, S.W. Carbocyclic 5′-Noruridine. Nucleosides Nucleotides Nucleic Acids 2000, 19, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.M.; Chen, X.; Schneller, S.W.; Ikeda, S.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E. Antiviral Enantiomeric Preference for 5′-Noraristeromycin. J. Med. Chem. 1994, 37, 551–554. [Google Scholar] [CrossRef]
- Jeong, L.S.; Yoo, S.J.; Lee, K.M.; Koo, M.J.; Choi, W.J.; Kim, H.O.; Moon, H.R.; Lee, M.Y.; Park, J.G.; Lee, S.K.; et al. Design, Synthesis, and Biological Evaluation of Fluoroneplanocin A as the Novel Mechanism-Based Inhibitor of S-Adenosylhomocysteine Hydrolase. J. Med. Chem. 2003, 46, 201–203. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W.; Chernousova, L.N.; Smirnova, T.G.; et al. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069. [Google Scholar] [CrossRef] [Green Version]
- Matyugina, E.; Novikov, M.; Babkov, D.; Ozerov, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Karpenko, I.; Chizhov, A.; Murthu, P.; et al. 5-Arylaminouracil Derivatives: New Inhibitors of Mycobacterium Tuberculosis. Chem. Biol. Drug Des. 2015, 86, 1387–1396. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Novikov, M.S.; Babkov, D.A.; Valuev-Elliston, V.T.; Vanpouille, C.; Zicari, S.; Corona, A.; Tramontano, E.; Margolis, L.B.; Khandazhinskaya, A.L.; et al. 5-Arylaminouracil Derivatives as Potential Dual-Action Agents. Acta Nat. 2015, 7, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, L.A.; Jasko, M.V.; Negrya, S.D.; Solyev, P.N.; Shevchenko, O.V.; Solodinin, A.P.; Kolonitskaya, D.P.; Karpenko, I.L.; Efremenkova, O.V.; Glukhova, A.A.; et al. Discovery of Novel N4-Alkylcytidines as Promising Antimicrobial Agents. Eur. J. Med. Chem. 2021, 215, 113212. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Shevchenko, O.V.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Negrya, S.D.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Avdanina, D.A.; et al. 3′-Amino Modifications Enhance the Antifungal Properties of N 4-Alkyl-5-Methylcytidines for Potential Biocides. New J. Chem. 2022, 46, 5614–5626. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J.; Nti-Addae, K.W. Prodrug Strategies to Overcome Poor Water Solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Khandazhinskaya, A.L.; Matyugina, E.S.; Shirokova, E.A. Anti-HIV therapy with AZT prodrugs: AZT phosphonate derivatives, current state and prospects. Expert Opin. Drug Metab. Toxicol. 2010, 6, 701–714. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Yasko, M.V.; Shirokova, E.A. The synthesis and antiviral properties of acyclic nucleoside analogues with a phosphonomethoxy fragment in the side chain. Curr. Med. Chem. 2006, 13, 2953–2980. [Google Scholar] [CrossRef]
- Jornada, D.; dos Santos Fernandes, G.; Chiba, D.; de Melo, T.; dos Santos, J.; Chung, M. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef] [Green Version]
- Mori, G.; Chiarelli, L.R.; Riccardi, G.; Pasca, M.R. New Prodrugs against Tuberculosis. Drug Discov. Today 2017, 22, 519–525. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Efremenkova, O.V.; Vasilyeva, B.F.; Sumarukova, I.G.; Kochetkov, S.N.; Alexandrova, L.A. Synthesis of Water-Soluble Prodrugs of 5-Modified 2′-Deoxyuridines and Their Antibacterial Activity. J. Antibiot. 2020, 73, 236–246. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Solyev, P.N.; Karpenko, I.L.; Shevchenko, O.V.; Chekhov, O.V.; Glukhova, A.A.; Vasilyeva, B.F.; Efimenko, T.A.; et al. Glycol and Phosphate Depot Forms of 4- and/or 5-Modified Nucleosides Exhibiting Antibacterial Activity. Mol. Biol. 2021, 55, 143–153. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Makarov, D.A.; Karpenko, I.L.; Solyev, P.N.; Chekhov, V.O.; Efremenkova, O.V.; Vasilieva, B.F.; Efimenko, T.A.; Kochetkov, S.N.; et al. Oligoglycol carbonate prodrugs of 5-modified 2′-deoxyuridines: Synthesis and antibacterial activity. Mendeleev Commun. 2022. In press. [Google Scholar]
- Bucher, H.C.; Griffith, L.E.; Guyatt, G.H.; Sudre, P.; Naef, M.; Sendi, P.; Battegay, M. Isoniazid Prophylaxis for Tuberculosis in HIV Infection: A Meta-Analysis of Randomized Controlled Trials. Aids 1999, 13, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Daley, C.L.; Small, P.M.; Schecter, G.F.; Schoolnik, G.K.; McAdam, R.A.; Jacobs, W.R.; Hopewell, P.C. An Outbreak of Tuberculosis with Accelerated Progression among Persons Infected with the Human Immunodeficiency Virus. N. Engl. J. Med. 1992, 326, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, A.; Jansson, M.; Sköld, M.; Rottenberg, M.E.; Källenius, G. Tuberculosis and HIV Co-Infection. PLoS Pathog. 2012, 8, e1002464. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, R.A.; Fangman, W.L. Yeast Gene CDC8 Encodes Thymidylate Kinase and Is Complemented by Herpes Thymidine Kinase Gene TK. Proc. Natl. Acad. Sci. USA 1984, 81, 5821–5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystal Structure of Inactive Mycobacterium Tuberculosis Thymidylate Kinase Complexed with Thymidine Monophosphate (TMP) at pH 4.6 (Resolution 1.85 Å). Available online: https://www.rcsb.org/structure/1N5I (accessed on 17 January 2022).
- Li de la Sierra, I.; Munier-Lehmann, H.; Gilles, A.; Bârzu, O.; Delarue, M. X-ray Structure of TMP Kinase from Mycobacterium tuberculosis Complexed with TMP at 1.95 Å Resolution. J. Mol. Biol. 2001, 311, 87–100. [Google Scholar] [CrossRef]
- Munier-Lehmann, H.; Chaffotte, A.; Pochet, S.; Labesse, G. Thymidylate Kinase of Mycobacterium tuberculosis: A Chimera Sharing Properties Common to Eukaryotic and Bacterial Enzymes. Protein Sci. 2001, 10, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Haouz, A.; Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Herdewijn, P.; Van Calenbergh, S.; Delarue, M. Enzymatic and Structural Analysis of Inhibitors Designed against Mycobacterium tuberculosis Thymidylate Kinase. J. Biol. Chem. 2003, 278, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Vanheusden, V.; Munier-Lehmann, H.; Pochet, S.; Herdewijn, P.; Van Calenbergh, S. Synthesis and Evaluation of Thymidine-5′-O-Monophosphate Analogues as Inhibitors of Mycobacterium tuberculosis Thymidylate Kinase. Bioorg. Med. Chem. Lett. 2002, 12, 2695–2698. [Google Scholar] [CrossRef]
- Fioravanti, E.; Adam, V.; Munier-Lehmann, H.; Bourgeois, D. The Crystal Structure of Mycobacterium Tuberculosis Thymidylate Kinase in Complex with 3’-Azidodeoxythymidine Monophosphate Suggests a Mechanism for Competitive Inhibition. Biochemistry 2005, 44, 130–137. [Google Scholar] [CrossRef]
- Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Dugué, L.; Heyerick, A.; De Keukeleire, D.; Pochet, S.; Busson, R.; Herdewijn, P.; Van Calenbergh, S. 3′-C-Branched-Chain-Substituted Nucleosides and Nucleotides as Potent Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. J. Med. Chem. 2003, 46, 3811–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanheusden, V.; Munier-Lehmann, H.; Froeyen, M.; Busson, R.; Rozenski, J.; Herdewijn, P.; Van Calenbergh, S. Discovery of Bicyclic Thymidine Analogues as Selective and High-Affinity Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. J. Med. Chem. 2004, 47, 6187–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pochet, S.; Dugue, L.; Douguet, D.; Labesse, G.; Munier-Lehmann, H. Nucleoside Analogues as Inhibitors of Thymidylate Kinases: Possible Therapeutic Applications. ChemBioChem 2002, 3, 108–110. [Google Scholar] [CrossRef]
- Pochet, S.; Dugué, L.; Labesse, G.; Delepierre, M.; Munier-Lehmann, H. Comparative Study of Purine and Pyrimidine Nucleoside Analogues Acting on the Thymidylate Kinases of Mycobacterium tuberculosis and of Humans. ChemBioChem 2003, 4, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Van Daele, I.; Munier-Lehmann, H.; Hendrickx, P.M.S.; Marchal, G.; Chavarot, P.; Froeyen, M.; Qing, L.; Martins, J.C.; Van Calenbergh, S. Synthesis and Biological Evaluation of Bicyclic Nucleosides as Inhibitors OfM. Tuberculosis Thymidylate Kinase. ChemMedChem 2006, 1, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, I.; Munier-Lehmann, H.; Froeyen, M.; Balzarini, J.; Van Calenbergh, S. Rational Design of 5′-Thiourea-Substituted α-Thymidine Analogues as Thymidine Monophosphate Kinase Inhibitors Capable of Inhibiting Mycobacterial Growth. J. Med. Chem. 2007, 50, 5281–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasse, C.; Huteau, V.; Douguet, D.; Munier-Lehmann, H.; Pochet, S. A New Family of Inhibitors of Mycobacterium tuberculosis Thymidine Monophosphate Kinase. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1057–1061. [Google Scholar] [CrossRef]
- Gasse, C.; Douguet, D.; Huteau, V.; Marchal, G.; Munier-Lehmann, H.; Pochet, S. Substituted Benzyl-Pyrimidines Targeting Thymidine Monophosphate Kinase of Mycobacterium tuberculosis: Synthesis and in Vitro Anti-Mycobacterial Activity. Bioorg. Med. Chem. 2008, 16, 6075–6085. [Google Scholar] [CrossRef]
- Familiar, O.; Munier-Lehmann, H.; Negri, A.; Gago, F.; Douguet, D.; Rigouts, L.; Hernández, A.; Camarasa, M.; Pérez-Pérez, M. Exploring Acyclic Nucleoside Analogues as Inhibitors of Mycobacterium tuberculosis Thymidylate Kinase. ChemMedChem 2008, 3, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Familiar, O.; Munier-Lehmann, H.; Aínsa, J.A.; Camarasa, M.-J.; Pérez-Pérez, M.-J. Design, Synthesis and Inhibitory Activity against Mycobacterium tuberculosis Thymidine Monophosphate Kinase of Acyclic Nucleoside Analogues with a Distal Imidazoquinolinone. Eur. J. Med. Chem. 2010, 45, 5910–5918. [Google Scholar] [CrossRef]
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Myllykallio, H.; Lipowski, G.; Leduc, D.; Filee, J.; Forterre, P.; Liebl, U. An Alternative Flavin-Dependent Mechanism for Thymidylate Synthesis. Science 2002, 297, 105–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehn, E.M.; Fleischmann, T.; Conrad, J.A.; Palfey, B.A.; Lesley, S.A.; Mathews, I.I.; Kohen, A. An Unusual Mechanism of Thymidylate Biosynthesis in Organisms Containing the ThyX Gene. Nature 2009, 458, 919–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziani, S.; Xia, Y.; Gurnon, J.R.; Van Etten, J.L.; Leduc, D.; Skouloubris, S.; Myllykallio, H.; Liebl, U. Functional Analysis of FAD-Dependent Thymidylate Synthase ThyX from Paramecium Bursaria Chlorella Virus-1. J. Biol. Chem. 2004, 279, 54340–54347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, I.I.; Deacon, A.M.; Canaves, J.M.; McMullan, D.; Lesley, S.A.; Agarwalla, S.; Kuhn, P. Functional Analysis of Substrate and Cofactor Complex Structures of a Thymidylate Synthase-Complementing Protein. Structure 2003, 11, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Karunaratne, K.; Kohen, A. Flavin-Dependent Thymidylate Synthase as a New Antibiotic Target. Molecules 2016, 21, 654. [Google Scholar] [CrossRef] [Green Version]
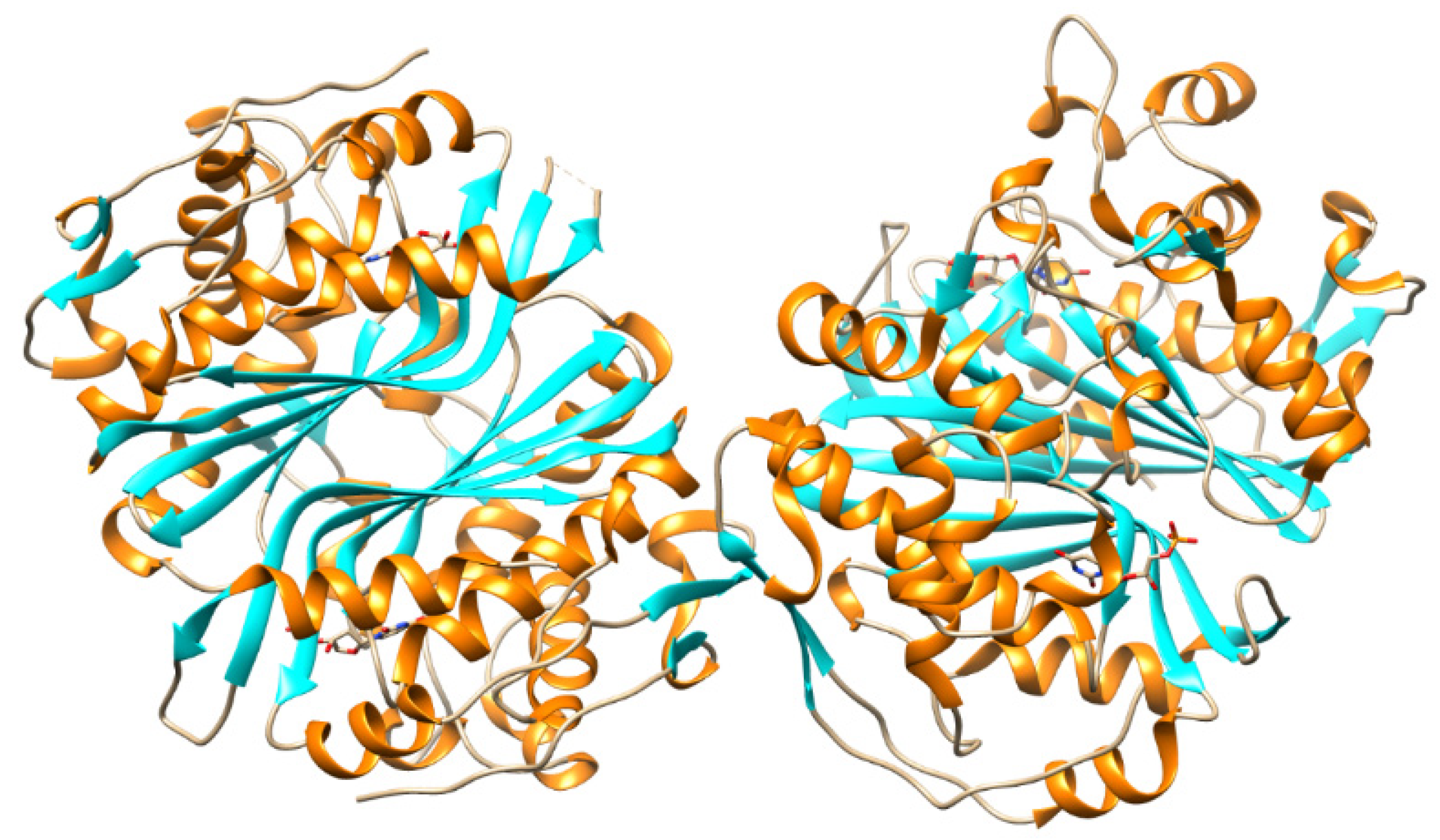
- Sampathkumar, P.; Turley, S.; Ulmer, J.E.; Rhie, H.G.; Sibley, C.H.; Hol, W.G.J. Structure of the Mycobacterium tuberculosis Flavin Dependent Thymidylate Synthase (MtbThyX) at 2.0Å Resolution. J. Mol. Biol. 2005, 352, 1091–1104. [Google Scholar] [CrossRef]
- Kögler, M.; Vanderhoydonck, B.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Herman, J.; Louat, T.; Parchina, A.; Sibley, C.; Lescrinier, E.; et al. Synthesis and Evaluation of 5-Substituted 2′-Deoxyuridine Monophosphate Analogues As Inhibitors of Flavin-Dependent Thymidylate Synthase in Mycobacterium tuberculosis. J. Med. Chem. 2011, 54, 4847–4862. [Google Scholar] [CrossRef] [Green Version]
- Crystal Structure of the Mtb ThyA in Complex with 5-Fluoro-DUMP. Available online: https://www.rcsb.org/structure/4FOA (accessed on 21 January 2022).
- Kögler, M.; Busson, R.; De Jonghe, S.; Rozenski, J.; Van Belle, K.; Louat, T.; Munier-Lehmann, H.; Herdewijn, P. Synthesis and Evaluation of 6-Aza-2′-Deoxyuridine Monophosphate Analogs as Inhibitors of Thymidylate Synthases, and as Substrates or Inhibitors of Thymidine Monophosphate Kinase in Mycobacterium tuberculosis. Chem. Biodivers. 2012, 9, 536–556. [Google Scholar] [CrossRef]
- Parchina, A.; Froeyen, M.; Margamuljana, L.; Rozenski, J.; De Jonghe, S.; Briers, Y.; Lavigne, R.; Herdewijn, P.; Lescrinier, E. Discovery of an Acyclic Nucleoside Phosphonate That Inhibits Mycobacterium tuberculosis ThyX Based on the Binding Mode of a 5-Alkynyl Substrate Analogue. ChemMedChem 2013, 8, 1373–1383. [Google Scholar] [CrossRef]
- McGuigan, C.; Derudas, M.; Gonczy, B.; Hinsinger, K.; Kandil, S.; Pertusati, F.; Serpi, M.; Snoeck, R.; Andrei, G.; Balzarini, J.; et al. ProTides of N-(3-(5-(2′-Deoxyuridine))Prop-2-Ynyl)Octanamide as Potential Anti-Tubercular and Anti-Viral Agents. Bioorg. Med. Chem. 2014, 22, 2816–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Murziani, P.; Slusarczyk, M.; Gonczy, B.; Vande Voorde, J.; Liekens, S.; Balzarini, J. Phosphoramidate ProTides of the Anticancer Agent FUDR Successfully Deliver the Preformed Bioactive Monophosphate in Cells and Confer Advantage over the Parent Nucleoside. J. Med. Chem. 2011, 54, 7247–7258. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef]
- McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A.; et al. Design, Synthesis and Evaluation of a Novel Double pro-Drug: INX-08189. A New Clinical Candidate for Hepatitis C Virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [Google Scholar] [CrossRef]
- Holy, A. Phosphonomethoxyalkyl Analogs of Nucleotides. Curr. Pharm. Des. 2003, 9, 2567–2592. [Google Scholar] [CrossRef]
- De Clercq, E.; Holý, A. Acyclic Nucleoside Phosphonates: A Key Class of Antiviral Drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Biteau, N.G.; Roy, V.; Lambry, J.-C.; Becker, H.F.; Myllykallio, H.; Agrofoglio, L.A. Synthesis of Acyclic Nucleoside Phosphonates Targeting Flavin-Dependent Thymidylate Synthase in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2021, 46, 116351. [Google Scholar] [CrossRef]
- Alexandrova, L.A.; Chekhov, V.O.; Shmalenyuk, E.R.; Kochetkov, S.N.; El-Asrar, R.A.; Herdewijn, P. Synthesis and Evaluation of C-5 Modified 2′-Deoxyuridine Monophosphates as Inhibitors of M. tuberculosis Thymidylate Synthase. Bioorg. Med. Chem. 2015, 23, 7131–7137. [Google Scholar] [CrossRef]
- Gibson, L.M.; Celeste, L.R.; Lovelace, L.L.; Lebioda, L. Structures of Human Thymidylate Synthase R163K with DUMP, FdUMP and Glutathione Show Asymmetric Ligand Binding. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 60–66. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). 2016. Available online: https://www.chemcomp.com/Products.htm (accessed on 21 January 2015).
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and Other Widely Available Force Fields for Conformational Energies and for Intermolecular-Interaction Energies and Geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
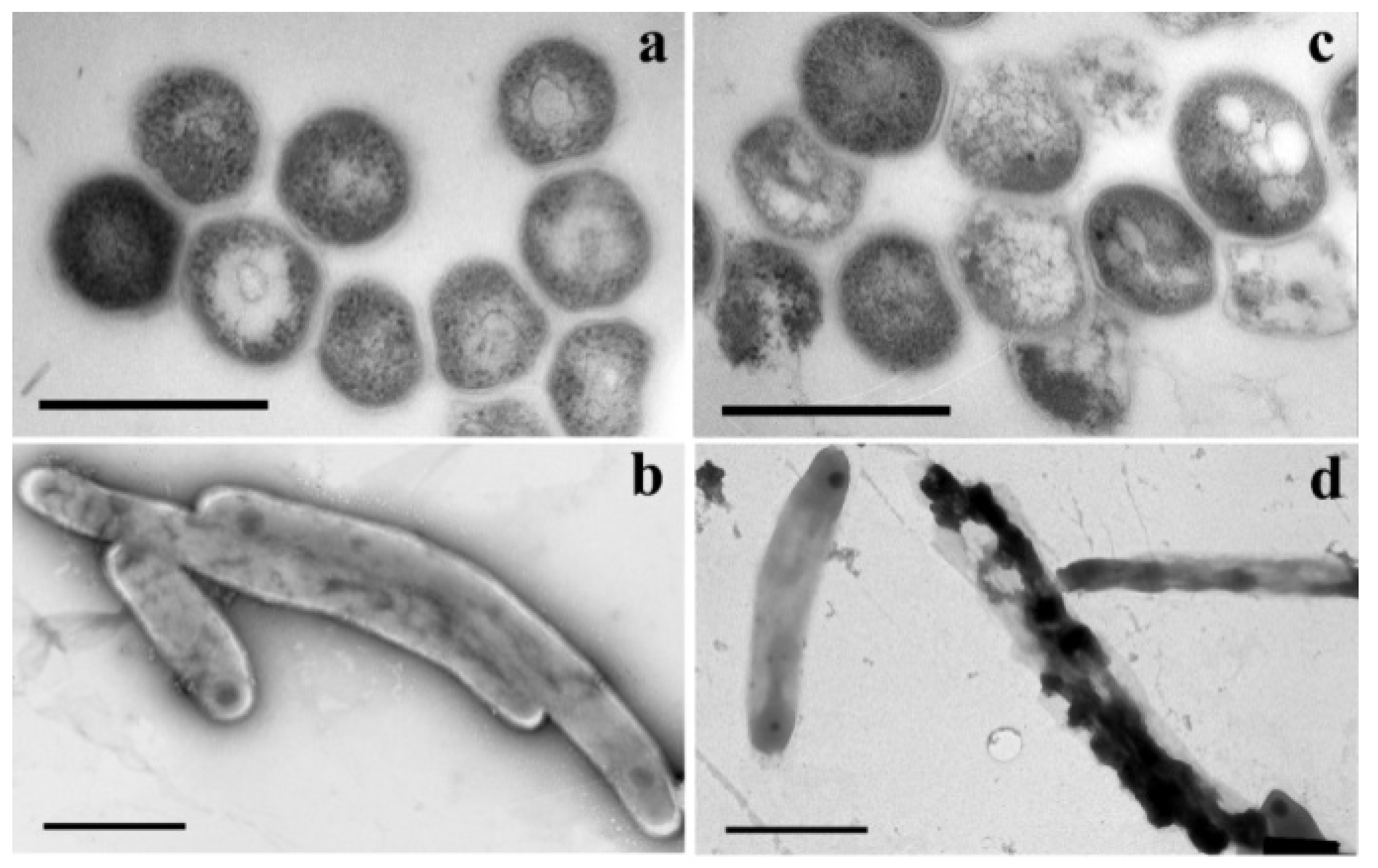
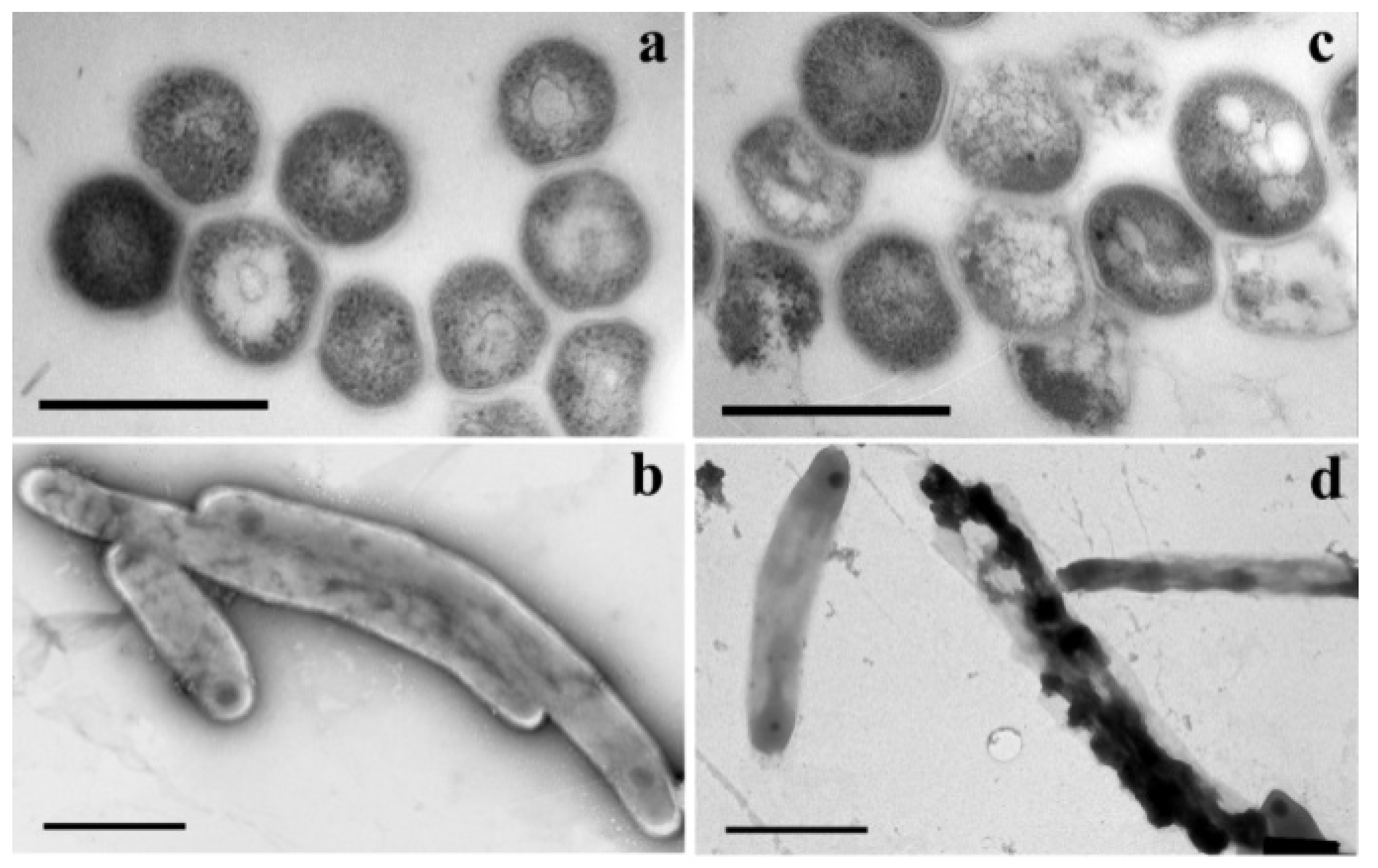
- Khandazhinskaya, A.L.; Matyugina, E.S.; Alexandrova, L.A.; Kezin, V.A.; Chernousova, L.N.; Smirnova, T.G.; Andreevskaya, S.N.; Popenko, V.I.; Leonova, O.G.; Kochetkov, S.N. Interaction of 5-Substituted Pyrimidine Nucleoside Analogues and M.tuberculosis: A View through an Electron Microscope. Biochimie 2020, 171–172, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Barisch, C.; Soldati, T. Mycobacterium Marinum Degrades Both Triacylglycerols and Phospholipids from Its Dictyostelium Host to Synthesise Its Own Triacylglycerols and Generate Lipid Inclusions. PLoS Pathog. 2017, 13, e1006095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, S.; Hai, H.T.; Thu, D.D.A.; Johnson, E.; Pielach, A.; Phu, N.H.; Thwaites, G.E.; Thuong, N.T.T. Ultrastructural Analysis of Cell Envelope and Accumulation of Lipid Inclusions in Clinical Mycobacterium tuberculosis Isolates from Sputum, Oxidative Stress, and Iron Deficiency. Front. Microbiol. 2018, 8, 2681. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffé, M.; Emile, J.-F.; Marchou, B.; Cardona, P.-J.; et al. Foamy Macrophages from Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. tuberculosis Persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Kayigire, X.A.; Friedrich, S.O.; van der Merwe, L.; Donald, P.R.; Diacon, A.H. Simultaneous Staining of Sputum Smears for Acid-Fast and Lipid-Containing Myobacterium tuberculosis Can Enhance the Clinical Evaluation of Antituberculosis Treatments. Tuberculosis 2015, 95, 770–779. [Google Scholar] [CrossRef]
- Velayati, A.A.; Farnia, P.; Ibrahim, T.A.; Haroun, R.Z.; Kuan, H.O.; Ghanavi, J.; Farnia, P.; Kabarei, A.N.; Tabarsi, P.; Omar, A.R.; et al. Differences in Cell Wall Thickness between Resistant and Nonresistant Strains of Mycobacterium tuberculosis: Using Transmission Electron Microscopy. Chemotherapy 2009, 55, 303–307. [Google Scholar] [CrossRef]
- Vijay, S.; Anand, D.; Ajitkumar, P. Unveiling Unusual Features of Formation of Septal Partition and Constriction in Mycobacteria—an Ultrastructural Study. J. Bacteriol. 2012, 194, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Seo, H.; Mahmud, H.A.; Islam, M.I.; Kim, Y.-S.; Lyu, J.; Nam, K.-W.; Lee, B.-E.; Lee, K.-I.; Song, H.-Y. In Vitro Effect of DFC-2 on Mycolic Acid Biosynthesis in Mycobacterium tuberculosis. J. Microbiol. Biotechnol. 2017, 27, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parumasivam, T.; Kumar, H.S.N.; Mohamad, S.; Ibrahim, P.; Sadikun, A. Effects of a Lipophilic Isoniazid Derivative on the Growth and Cellular Morphogenesis of Mycobacterium tuberculosis H37Rv. Int. J. Pharm. Pharm. Sci. 2013, 5, 43–50. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandrova, L.A.; Khandazhinskaya, A.L.; Matyugina, E.S.; Makarov, D.A.; Kochetkov, S.N. Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms 2022, 10, 1299. https://doi.org/10.3390/microorganisms10071299
Alexandrova LA, Khandazhinskaya AL, Matyugina ES, Makarov DA, Kochetkov SN. Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms. 2022; 10(7):1299. https://doi.org/10.3390/microorganisms10071299
Chicago/Turabian StyleAlexandrova, Liudmila A., Anastasia L. Khandazhinskaya, Elena S. Matyugina, Dmitriy A. Makarov, and Sergey N. Kochetkov. 2022. "Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors" Microorganisms 10, no. 7: 1299. https://doi.org/10.3390/microorganisms10071299
APA StyleAlexandrova, L. A., Khandazhinskaya, A. L., Matyugina, E. S., Makarov, D. A., & Kochetkov, S. N. (2022). Analogues of Pyrimidine Nucleosides as Mycobacteria Growth Inhibitors. Microorganisms, 10(7), 1299. https://doi.org/10.3390/microorganisms10071299