
Current Treatments to Control African Trypanosomiasis and One Health Perspective

 , , , ,
, , , ,  , and
, and
Abstract
:1. Epidemiology of African Trypanosomiasis
1.1. Human African Trypanosomiasis
1.2. Animal African Trypanosomiasis
2. The Disease
2.1. The Human Disease
2.2. The Animal Diseases
3. Current Treatments and Therapeutic Challenges for HAT
3.1. Pentamidine and Suramin
3.2. Melarsoprol
3.3. Eflornithine and NECT (Nifurtimox–Eflornithine Combination Therapy)
3.4. Nifurtimox (NFX)
3.5. Fexinidazole
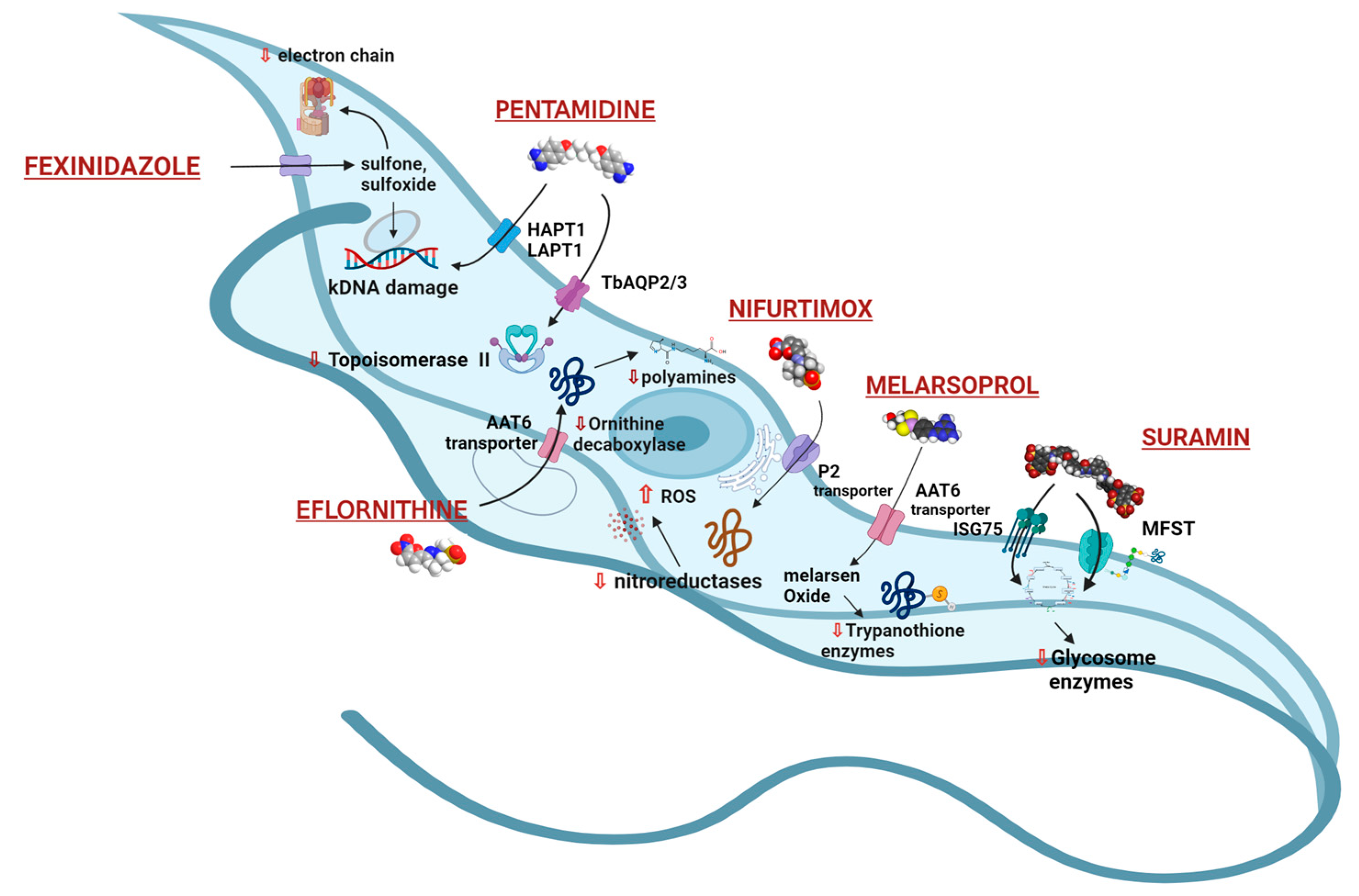

4. Current Treatments and Therapeutic Challenges for AAT
4.1. Phenanthridine (Homidium (or Ethidium Bromide), Isometamidium Chloride)
4.2. Aminoquinaldine (Quinapyramine)
4.3. Diamidine (Diminazene)
4.4. Melaminophenyl Arsenical
4.5. Suramin

5. Recent Clinical Candidate Drugs for HAT
5.1. Parafuramidine
5.2. Acoziborole
6. Recent Pharmaceutical Discovery and Development against African Trypanosomiasis
6.1. Nucleoside Analogues
6.2. New Molecules from Structure-Based Drug Design (SBDD)
6.3. Chemotypes Derived from High-Throughput Screening (HTS)
7. Drug Resistance in T. brucei
8. Surveillance and Disease Control Measures: Targets for African Trypanosomiasis Elimination under One Health Umbrella
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Masocha, W.; Kristensson, K. Human African Trypanosomiasis: How Do the Parasites Enter and Cause Dysfunctions of the Nervous System in Murine Models? Brain Res. Bull. 2019, 145, 18–29. [Google Scholar] [CrossRef]
- Black, S.J.; Seed, J.R.; Murphy, N.B. Innate and Acquired Resistance to African Trypanosomiasis. J. Parasitol. 2001, 87, 1. [Google Scholar] [CrossRef]
- WHO. Elimination of Human African Trypanosomiasis as Public Health Problem. Wkly. Epidemiol. Rec. 2021, 96, 176. [Google Scholar]
- WHO. Human African Trypanosomiasis (Sleeping Sickness). Available online: https://www.who.int/data/gho/data/themes/topics/human-african-trypanosomiasis (accessed on 8 March 2022).
- Kasozi, K.I.; Namayanja, M.; Gaithuma, A.K.; Mahero, M.; Matovu, E.; Yamagishi, J.; Sugimoto, C.; MacLeod, E. Prevalence of hemoprotozoan parasites in small ruminants along a human-livestock-wildlife interface in western Uganda. Vet. Parasitol. Reg. Stud. Rep. 2019, 17, 100309. [Google Scholar] [CrossRef]
- Yaro, M.; Munyard, K.A.; Stear, M.J.; Groth, D.M. Combatting African Animal Trypanosomiasis (AAT) in Livestock: The Potential Role of Trypanotolerance. Vet. Parasitol. 2016, 225, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Desquesnes, M.; Gonzatti, M.; Sazmand, A.; Thévenon, S.; Bossard, G.; Boulangé, A.; Gimonneau, G.; Truc, P.; Herder, S.; Ravel, S.; et al. A Review on the Diagnosis of Animal Trypanosomoses. Parasites Vectors 2022, 15, 64. [Google Scholar] [CrossRef]
- Isaac, C.; Ohiolei, J.A.; Ebhodaghe, F.; Igbinosa, I.B.; Eze, A.A. Animal African Trypanosomiasis in Nigeria: A Long Way from Elimination/Eradication. Acta Trop. 2017, 176, 323–331. [Google Scholar] [CrossRef]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African Trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; Rodgers, J. Clinical and Neuropathogenetic Aspects of Human African Trypanosomiasis. Front. Immunol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- WHO. Number of New Reported Cases of Human African Trypanosomiasis (T. b. rhodesiense). Available online: https://www.who.int/data/gho/data/indicators/indicator-details/gho/number-of-new-reported-cases-of-human-african-trypanosomiasis-(t-b-rhodesiense) (accessed on 25 March 2022).
- Pays, E.; Nolan, D.P. Genetic and Immunological Basis of Human African Trypanosomiasis. Curr. Opin. Immunol. 2021, 72, 13–20. [Google Scholar] [CrossRef]
- Udensi, U.K.; Fagbenro-Beyioku, A.F. Effect of Ivermectin on Trypanosoma brucei brucei in Experimentally Infected Mice. J. Vector Borne Dis. 2012, 49, 143–150. [Google Scholar]
- Checchi, F.; Felipe, J.A.N.; Haydon, D.T.; Chandramohan, D.; Chappuis, F. Estimates of the Duration of the Early and Late Stage of Gambiense Sleeping Sickness. BMC Infect. Dis. 2008, 8, 16. [Google Scholar] [CrossRef]
- CDC. Diseases: Neglected Tropical Diseases; Centers for Disease Control: Altanta, GA, USA, 2020. Available online: https://www.cdc.gov/globalhealth/ntd/index.html (accessed on 30 March 2022).
- European Medicine Agency (EMA); Committee for Veterinary Medicinal Products. Advice on the Designation of Antimicrobials or Groups of Antimicrobials Reserved for Treatment of Certain Infections in Humans; EMA: Amsterdam, The Netherlands, 2022.
- Chitanga, S.; Marcotty, T.; Namangala, B.; Van Den Bossche, P.; Van Den Abbeele, J.; Delespaux, V. High Prevalence of Drug Resistance in Animal Trypanosomes without a History of Drug Exposure. PLoS Negl. Trop. Dis. 2011, 5, E1454. [Google Scholar] [CrossRef] [Green Version]
- Dickie, E.; Giordani, F.; Gould, M.K.; Mäser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Infect. Dis. 2020, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, J.S.; Wortmann, G.W.; Kirchhoff, L.V. Drugs for Protozoal Infections other than Malaria. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 42, pp. 535–543.e3. [Google Scholar]
- Thomas, J.A.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into Antitrypanosomal Drug Mode-of-Action from Cytology-Based Profiling. PLoS Negl. Trop. Dis. 2018, 12, E0006980. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug Resistance in African Trypanosomiasis: The Melarsoprol and Pentamidine Story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Gill, B.; Malhotra, M. Prophylactic Activity of Suramin Complexes in ‘Surra’ (Trypanosoma evansi). Nature 1963, 200, 285–286. [Google Scholar] [CrossRef]
- Zoltner, M.; Campagnaro, G.D.; Taleva, G.; Burrell, A.; Cerone, M.; Leung, K.F.; Achcar, F.; Horn, D.; Vaughan, S.; Gadelha, C.; et al. Suramin Exposure Alters Cellular Metabolism and Mitochondrial Energy Production in African Trypanosomes. J. Biol. Chem. 2020, 295, 8331–8347. [Google Scholar] [CrossRef]
- Li, M.; Gaussmann, S.; Tippler, B.; Ott, J.; Popowicz, G.M.; Schliebs, W.; Sattler, M.; Erdmann, R.; Kalel, V.C. Novel Trypanocidal Inhibitors that Block Glycosome Biogenesis by Targeting PEX3–PEX19 Interaction. Front. Cell Dev. Biol. 2021, 9, 737159. [Google Scholar] [CrossRef]
- Kennedy, P.G. The Continuing Problem of Human African Trypanosomiasis (Sleeping Sickness). Ann. Neurol. 2008, 64, 116–126. [Google Scholar] [CrossRef]
- Kennedy, P.G. Human African Trypanosomiasis of the CNS: Current Issues and Challenges. J. Clin. Investig. 2004, 113, 496–504. [Google Scholar] [CrossRef]
- Kennedy, P.G. Clinical Features, Diagnosis, and Treatment of Human African Trypanosomiasis (Sleeping Sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Atouguia, J.L.M.; Kennedy, P.G. Neurological Aspects of Human African Trypanosomiasis. In Infectious Diseases of the Nervous System; Davies, L.E., Kennedy, P.G.E., Eds.; Butterworth-Heinemann: Oxford, UK, 2000; pp. 321–372. ISBN 0750642130. [Google Scholar]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A Current Analysis of Chemotherapy Strategies for the Treatment of Human African Trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, S.C.; Nerima, B.; Mäser, P.; Brun, R. Melarsoprol- and Pentamidine-Resistant Trypanosoma brucei rhodesiense Populations and Their Cross-Resistance. Int. J. Parasitol. 2007, 37, 1443–1448. [Google Scholar] [CrossRef]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic Drug Resistance Mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.; Barrett, M.P. A Molecular Mechanism for Eflornithine Resistance in African Trypanosomes. PLoS Pathog. 2010, 6, E1001204. [Google Scholar] [CrossRef] [Green Version]
- Priotto, G.; Pinoges, L.; Fursa, I.B.; Burke, B.; Nicolay, N.; Grillet, G.; Hewison, C.; Balasegaram, M. Safety and Effectiveness of First Line Eflornithine for Trypanosoma brucei gambiense Sleeping Sickness in Sudan: Cohort Study. BMJ 2008, 336, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, K.; Paulino, M.; Salas, C.O.; Zarate-Ramos, J.J.; Vera, B.; Rivera, G. Trypanothione Reductase: A Target for the Development of Anti-Trypanosoma cruzi Drugs. Mini-Rev. Med. Chem. 2017, 17, 939–946. [Google Scholar] [CrossRef]
- Lindner, A.K.; Lejon, V.; Chappuis, F.; Seixas, J.; Kazumba, L.; Barrett, M.P.; Mwamba, E.; Erphas, O.; Akl, E.A.; Villanueva, G.; et al. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020, 20, e38–e46. [Google Scholar] [CrossRef]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Pollastri, M.P. Fexinidazole: A New Drug for African Sleeping Sickness on the Horizon. Trends Parasitol. 2018, 34, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of Fexinidazole in Patients with Stage 1 or Early Stage 2 Human African Trypanosomiasis (HAT) Due to T. b. gambiense: A Prospective, Multicentre, Open-Label Cohort Study, Plug-in to the Pivotal Study. Available online: https://clinicaltrials.gov/ct2/show/NCT02169557 (accessed on 20 April 2022).
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Torreele, E.; Trunz, B.B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole: A New Oral Nitroimidazole Drug Candidate Entering Clinical Development for the Treatment of Sleeping Sickness. PLoS Negl. Trop. Dis. 2010, 4, E923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, G.; Orta, J.F.; Sharma, M.; Mukhopadhyay, R. Trypanosomatid Aquaporins: Roles in Physiology and Drug Response. Diseases 2014, 2, 3–23. [Google Scholar] [CrossRef] [Green Version]
- De Koning, P.H. The Drugs of Sleeping Sickness: Their Mechanisms of Action and Resistance, and a Brief History. Trop. Med. Infect. Dis. 2020, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Zoltner, M.; Horn, D.; De Koning, H.P.; Field, M.C. Exploiting the Achilles’ Heel of Membrane Trafficking in Trypanosomes. Curr. Opin. Microbiol. 2016, 34, 97–103. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.P. Uptake of Pentamidine in Trypanosoma brucei brucei Is Mediated by Three Distinct Transporters: Implications for Cross-Resistance with Arsenicals. Mol. Pharmacol. 2001, 59, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Andrés, D.A.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef]
- Kazibwe, A.J.; Nerima, B.; de Koning, H.P.; Mäser, P.; Barrett, M.P.; Matovu, E. Genotypic status of the TbAT1/P2 adenosine transporter of Trypanosoma brucei gambiense isolates from Northwestern Uganda following melarsoprol withdrawal. PLoS Negl. Trop. Dis. 2009, 3, e523. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.; Argaw, D. Monitoring the Elimination of Human African Trypanosomiasis: Update to 2016. PLoS Negl. Trop. Dis. 2018, 12, E0006890. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of Human African Trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef] [PubMed]
- FAO. African Animal Trypanosomiasis. Available online: https://www.fao.org/3/ah809e/ah809e02.htm (accessed on 25 March 2022).
- Richards, S.; Morrison, L.J.; Torr, S.J.; Barrett, M.P.; Manangwa, O.; Mramba, F.; Auty, H. Pharma to Farmer: Field Challenges of Optimizing Trypanocide Use in African Animal Trypanosomiasis. Trends Parasitol. 2021, 37, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.S.; Engel, J.A.; Chua, M.J.; Fisher, G.M.; Skinner-Adams, T.S.; Andrews, K.T. Adaptation of the [3H]Hypoxanthine Uptake Assay for In Vitro-Cultured Plasmodium knowlesi Malaria Parasites. Antimicrob. Agents Chemother. 2016, 60, 4361–4363. [Google Scholar] [CrossRef] [Green Version]
- Eisler, M.C.; Stevenson, P.; Munga, L.; Smyth, J.B. Concentrations of Isometamidium Chloride (Samorin) in Sera of Zebu Cattle which Showed Evidence of Hepatotoxicity Following Frequent Trypanocidal Treatments. J. Vet. Pharmacol. Ther. 1997, 20, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Assefa, S.; Shibeshi, W. Drug Resistance in African Animal Trypanosomes: A Review. Afr. J. Microbiol. Res. 2018, 12, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, M. Dyes, Trypanosomiasis and DNA: A Historical and Critical Review. Biotech. Histochem. 2010, 85, 341–354. [Google Scholar] [CrossRef]
- Chowdhury, R.A.; Bakshi, R.; Wang, J.; Yildirir, G.; Liu, B.; Pappas-Brown, V.; Tolun, G.; Griffith, J.D.; Shapiro, T.A.; Jensen, R.E.; et al. The Killing of African Trypanosomes by Ethidium Bromide. PLoS Pathog. 2010, 6, E1001226. [Google Scholar] [CrossRef] [Green Version]
- Büscher, P.; Gonzatti, M.I.; Hébert, L.; Inoue, N.; Pascucci, I.; Schnaufer, A.; Suganuma, K.; Touratier, L.; Van Reet, N. Equine Trypanosomosis: Enigmas and Diagnostic Challenges. Parasites Vectors 2019, 12, 234. [Google Scholar] [CrossRef]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; de Koning, H.P.; Barrett, M.P. The Animal Trypanosomiases and Their Chemotherapy: A Review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Kuriakose, S.; Muleme, H.; Onyilagha, C.; Okeke, E.; Uzonna, J.E. Diminazene Aceturate (Berenil) Modulates LPS Induced pro-Inflammatory Cytokine Production by Inhibiting Phosphorylation of MAPKs and STAT Proteins. Innate Immun. 2014, 20, 760–773. [Google Scholar] [CrossRef]
- Raynaud, J.P. Thiacetarsamide (Adulticide) versus Melarsomine (RM340) Developed as Macrofilaricide (Adulticide and Larvicide) to Cure Canine Heartworm Infection in Dogs. Ann. Rech. Vet. 1992, 23, 1–25. [Google Scholar] [PubMed]
- Kazosi, K.I.; MacLeod, E.T.; Ntulume, I.; Welburn, S.C. An Update on African Trypanocide Pharmaceutics and Resistance. Front. Vet. Sci. 2022, 9, 828111. [Google Scholar] [CrossRef]
- Moore, T.A. 246e: Agents Used to Treat Parasitic Infections. In Harrison’s Principles of Internal Medicine, 19th ed.; Kasper, D., Fauci, A., Hauser, S., Longo, D., Jameson, J.L., Loscalzo, J., Eds.; McGraw-Hill: New York, NY, USA, 2014. [Google Scholar]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, G.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G Protein-Coupled Nucleotide Receptor: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef]
- Stewart, M.L.; Bueno, G.J.; Baliani, A.; Klenke, B.; Brun, R.; Brock, J.M.; Gilbert, I.H.; Barrett, M.P. Trypanocidal Activity of Melamine Based Nitroheterocycles. Antimicrob. Agents Chemother. 2004, 48, 1733–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug Resistance in Human African Trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-Throughput Decoding of Antitrypanosomal Drug Efficacy and Resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, S.; Patterson, S.; Stojanovski, L.; Simeons, F.R.C.; Norval, S.; Kime, R.; Read, K.D.; Fairlamb, A.H. The Anti-Trypanosome Drug Fexinidazole Shows Potential for Treating Visceral Leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. [Google Scholar] [CrossRef] [Green Version]
- Mdachi, R.E.; Thuita, J.K.; Kagira, J.M.; Ngotho, J.M.; Murilla, G.A.; Ndung’u, J.M.; Tidwell, R.R.; Hall, J.E.; Brun, R. Efficacy of the Novel Diamidine Compound 2,5-Bis(4-Amidinophenyl)- Furan-Bis-O-Methlylamidoxime (Pafuramidine, DB289) against Trypanosoma brucei rhodesiense infection in vervet monkeys after oral administration. Antimicrob. Agents Chemother. 2009, 53, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Begolo, D.; Vincent, I.M.; Giordani, F.; Pöhner, I.; Witty, M.J.; Rowan, T.G.; Bengaly, Z.; Gillingwater, K.; Freund, Y.; Wade, R.C.; et al. The trypanocidal benzoxaborole AN7973 inhibits trypanosome mRNA processing. PLoS Pathog. 2018, 14, e1007315. [Google Scholar] [CrossRef] [Green Version]
- Wall, J.R.; Rico, E.; Lukac, I.; Zuccotto, F.; Elg, S.; Gilbert, I.H.; Freund, Y.; Alley, M.R.K.; Field, M.C.; Wyllie, S.; et al. Clinical and Veterinary Trypanocidal Benzoxaboroles Target CPSF3. Proc. Natl. Acad. Sci. USA 2018, 115, 9616–9621. [Google Scholar] [CrossRef] [Green Version]
- Steketee, P.C.; Vincent, I.M.; Achcar, F.; Giordani, F.; Kim, D.H.; Creek, D.J.; Freund, Y.; Jacobs, R.; Rattigan, K.; Horn, D.; et al. Benzoxaborole Treatment Perturbs S-Adenosyl-L-Methionine Metabolism in Trypanosoma brucei. PLoS Negl. Trop. Dis. 2018, 12, e0006450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasozi, K.I.; Zirintunda, G.; Ssempijja, F.; Buyinza, B.; Alzahrani, K.J.; Matama, K.; Nakimbugwe, H.N.; Alkazmi, L.M.; Onanyang, D.; Bogere, P.; et al. Epidemiology of Trypanosomiasis in Wildlife—Implications for Humans at the Wildlife Interface in Africa. Front. Vet. Sci. 2021, 8, 621699. [Google Scholar] [CrossRef]
- Safety and Tolerability Study of Acoziborole in G-HAT Seropositive Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT05256017 (accessed on 4 April 2022).
- Hammond, D.J.; Gutteridge, W.E. Purine and Pyrimidine Metabolism in the Trypanosomatidae. Mol. Biochem. Parasitol. 1984, 13, 243–261. [Google Scholar] [CrossRef]
- Boitz, J.M.; Ullman, B.; Jardim, A.; Carter, N.S. Purine Salvage in Leishmania: Complex or Simple by Design? Trends Parasitol. 2012, 28, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Aoki, J.I.; Coelho, A.C.; Muxel, S.M.; Zampieri, R.A.; Ramos Sanchez, E.M.; Nerland, A.H.; Floeter-Winter, L.M.; Cotrim, P.C. Characterization of a Novel Endoplasmic Reticulum Protein Involved in Tubercidin Resistance in Leishmania major. PLoS Negl. Trop. Dis. 2016, 10, e0004972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, J. Cordycepin, an Antitumour Antibiotic with Trypanocidal Properties. Trans. R. Soc. Trop. Med. Hyg. 1966, 60, 8. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Lundbäck, T.; Yeheskieli, E.; Sjöberg, B.; Gustavsson, A.-L.; Svensson, R.; Olivera, G.C.; Eze, A.A.; de Koning, H.P.; Hammarström, L.G.J.; et al. Structure–Activity Relationships of Synthetic Cordycepin Analogues as Experimental Therapeutics for African trypanosomiasis. J. Med. Chem. 2013, 56, 9861–9873. [Google Scholar] [CrossRef]
- Peña, I.; Manzano, M.P.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 8771. [Google Scholar] [CrossRef] [Green Version]
- Hulpia, F.; Campagnaro, G.D.; Scortichini, M.; Van Hecke, K.; Maes, L.; de Koning, H.P.; Caljon, G.; Van Calenbergh, S. Revisiting Tubercidin against Kinetoplastid Parasites: Aromatic Substitutions at Position 7 Improve Activity and Reduce Toxicity. Eur. J. Med. Chem. 2019, 164, 689–705. [Google Scholar] [CrossRef]
- Hulpia, F.; Mabille, D.; Campagnaro, G.D.; Schumann, G.; Maes, L.; Roditi, I.; Hofer, A.; de Koning, H.P.; Caljon, G.; Van Calenbergh, S. Combining Tubercidin and Cordycepin Scaffolds Results in Highly Active Candidates to Treat Late-Stage Sleeping Sickness. Nat. Commun. 2019, 10, 5564. [Google Scholar] [CrossRef] [Green Version]
- Blaazer, A.R.; Singh, A.K.; de Heuvel, E.; Edink, E.; Orrling, K.M.; Veerman, J.J.N.; Bergh, T.V.D.; Jansen, C.; Balasubramaniam, E.; Mooij, W.J.; et al. Targeting a subpocket in Trypanosoma brucei phosphodiesterase B1 (TbrPDEB1) enables the structure-based discovery of selective inhibitors with trypanocidal activity. J. Med. Chem. 2018, 61, 3870–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poehner, I.; Quotadamo, A.; Panecka-Hofman, J.; Luciani, R.; Santucci, M.; Linciano, P.; Landi, G.; di Pisa, F.; dello Iacono, L.; Pozzi, C.; et al. Multitarget, Selective Compound Design Yields Picomolar Inhibitors of a Kinetoplastid Pteridine Reductase 1. J. Med. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.T.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.S.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Spinks, D.; Smith, V.; Thompson, S.; Robinson, D.A.; Luksch, T.; Smith, A.; Torrie, L.S.; McElroy, S.; Stojanovski, L.; Norval, S.; et al. Development of Small-Molecule Trypanosoma brucei N-Myristoyltransferase Inhibitors: Discovery and Optimisation of a Novel Binding Mode. ChemMedChem 2015, 10, 1821–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klug, D.M.; Mavrogiannaki, E.M.; Forbes, K.C.; Silva, L.; Diaz-Gonzalez, R.; Pérez-Moreno, G.; Ceballos-Pérez, G.; Garcia-Hernández, R.; Bosch-Navarrete, C.; Cordón-Obras, C.; et al. Lead Optimization of 3,5-Disubstituted-7-Azaindoles for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2021, 64, 9404–9430. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Diaz-Gonzalez, R.; Ceballos-Perez, G.; Rojas-Barros, D.I.; Gunaganti, N.; Gillingwater, K.; Martinez-Martinez, M.S.; Manzano, P.; Navarro, M.; Pollastri, M.P. Medicinal Chemistry Optimization of a Diaminopurine Chemotype: Toward a Lead for Trypanosoma brucei Inhibitors. J. Med. Chem. 2020, 63, 9912–9927. [Google Scholar] [CrossRef] [PubMed]
- Tassone, G.; Landi, G.; Linciano, P.; Francesconi, V.; Tonelli, M.; Tagliazucchi, L.; Costi, M.P.; Mangani, S.; Pozzi, C. Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase. Pharmaceuticals 2021, 14, 636. [Google Scholar] [CrossRef]
- Gashumba, J.K.; Komba, E.K.; Truc, P.; Allingham, R.M.; Ferris, V.; Godfrey, D.G. The Persistence of Genetic Homogeneity among Trypanosoma brucei rhodesiense Isolates from Patients in North-West Tanzania. Acta Trop. 1994, 56, 341–348. [Google Scholar] [CrossRef]
- Tihon, E.; Imamura, H.; Van Den Broeck, F.; Vermeiren, L.; Dujardin, J.C.; Van Den Abbeele, J. Genomic analysis of Isometamidium Chloride resistance in Trypanosoma congolense. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 350–361. [Google Scholar] [CrossRef]
- De Koning, H.P. Ever-Increasing Complexities of Diamidine and Arsenical Crossresistance in African Trypanosomes. Trends Parasitol. 2008, 24, 345–349. [Google Scholar] [CrossRef]
- Wilkes, J.M.; Mulugeta, W.; Wells, C.; Peregrine, A.S. Modulation of Mitochondrial Electrical Potential: A Candidate Mechanism for Drug Resistance in African Trypanosomes. Biochem. J. 1997, 326 Pt 3, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, S.; Gould, M.K.; Dewar, C.E.; Schnaufer, A.C. Single Point Mutations in ATP Synthase Compensate for Mitochondrial Genome Loss in Trypanosomes. Proc. Natl. Acad. Sci. USA 2013, 110, 14741–14746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okello, A.L.; Bardosh, K.; Smith, J.; Welburn, S.C. One Health: Past Successes and Future Challenges in Three African Contexts. PLoS Negl. Trop. Dis. 2014, 8, E2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamill, L.; Picozzi, K.; Fyfe, J.; Von Wissmann, B.; Wastling, S.; Wardrop, N.; Selby, R.; Acup, C.A.; Bardosh, K.L.; Muhanguzi, D.; et al. Evaluating the impact of targeting livestock for the prevention of human and animal trypanosomiasis, at village level, in districts newly affected with T. b. rhodesiense in Uganda. Infect. Dis. Poverty 2017, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Elimination of Human African Trypanosomiasis as Public Health Problem. Wkly. Epidemiol. Rec. 2021. Available online: https://www.cdc.gov/parasites/sleepingsickness/health_professionals/index.html#dx (accessed on 25 March 2022).
- Waiswa, C.; Azuba, R.; Makeba, J.; Waiswa, I.C.; Wangoola, R.M. Experiences of the one-health approach by the Uganda Trypanosomiasis Control Council and its secretariat in the control of zoonotic sleeping sickness in Uganda. Parasite Epidemiol. Control 2020, 11, e00185. [Google Scholar] [CrossRef] [PubMed]
- Picado, A.; Ndung’u, J. Elimination of Sleeping Sickness in Uganda Could Be Jeopardised by Conflict in South Sudan. Lancet 2017, 5, E28–E29. [Google Scholar] [CrossRef] [Green Version]
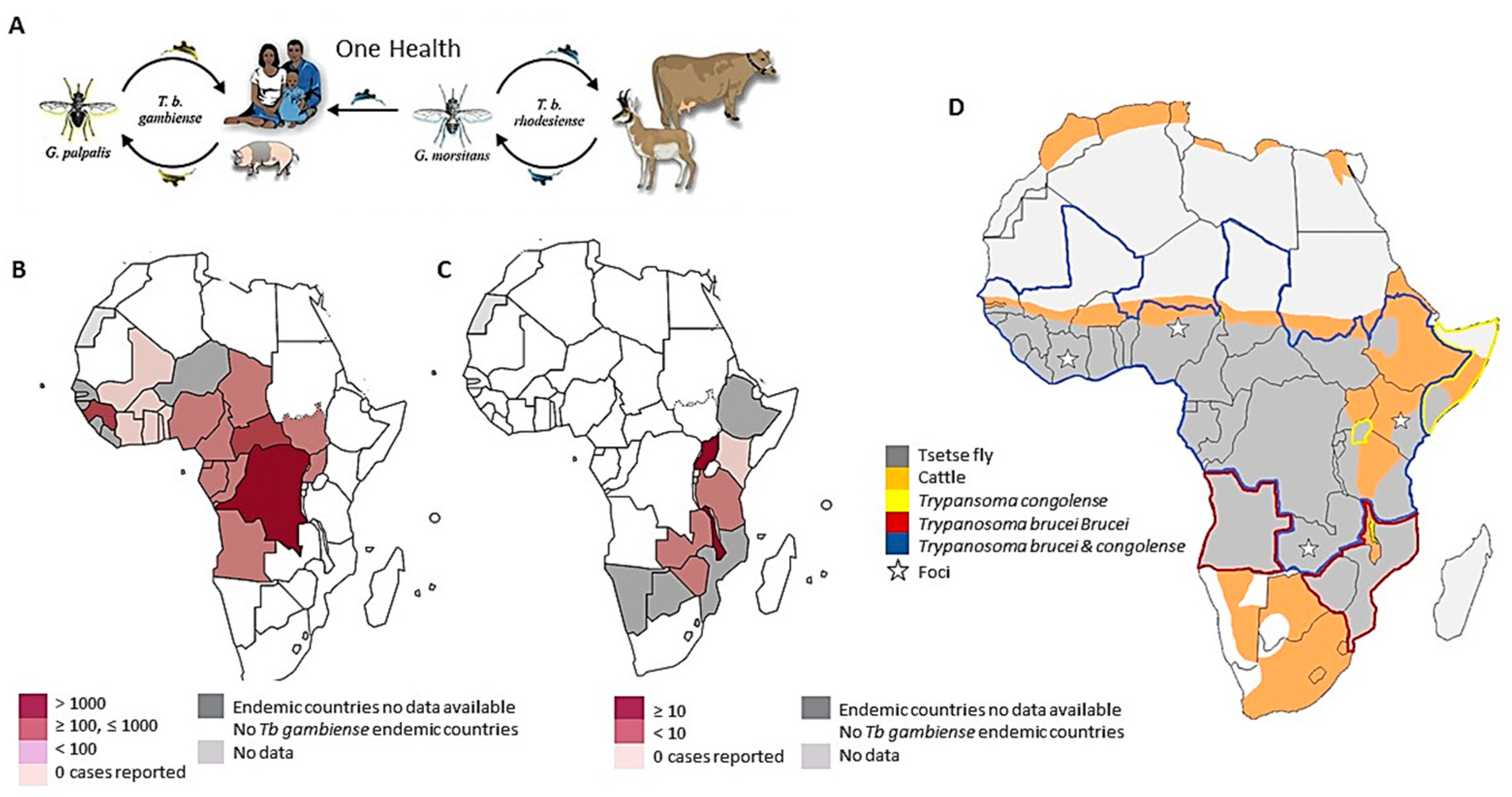

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approved Drugs for HAT | Species | Dose |
|---|---|---|
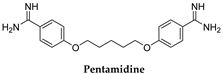 | T. b. gambiense | 4 mg/kg/day IM × 10 days |
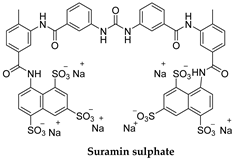 | T. b. rhodesiense T. b. gambiense | Test dose of 4–5 mg/kg (day 1), then 20 mg/kg, weekly ×5 weeks (maximal dose/injection: 1 g) |
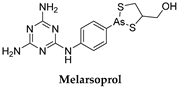 | T. b. rhodesiense | 2.2 mg/kg/day × 10 days, usually accompanied by prednisolone1 mg/kg/day |
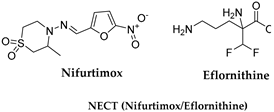 | T. b. gambiense | Nifurtimox: 15 mg/kg 24 h, 3 dose × 10 days Eflornithine: 400 mg/kg/day 2-h infusion × 7 days |
 | T. b. gambiense | For patients 20–35 kg; 1200 mg/day × 4 days and then 600 mg/day × 6 days For patients ≥ 35 kg; 1800 mg/day × 4 days and then 1200 mg/day × 6 days |
| Drugs in Current Treatment for AAT | Trypanosome Species Target | Animal Species | Applicability |
|---|---|---|---|
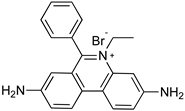 | T. vivax T. congolense T. b. | Cattle Sheep Goats | Treatment |
| Homidium bromide | |||
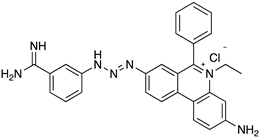 | T. vivax T. congolense T. b. | Cattle Sheep Goats Horses | Treatment or prophylaxis |
| Isometamidium chloride | |||
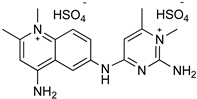 | T. b. T. evansi | Horses Camels Cattle | Prophylaxis |
| Quinapyramine sulphate | |||
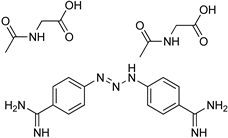 | T. congolense T. evansi | Cattle Sheep Goats Dogs | Treatment |
| Diminazene aceturate | |||
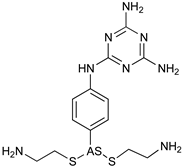 | T. evansi | Camels | Treatment |
| Melarsomine |
| Parafuramidine, Furamidine and Aza-Derivatives | |
|---|---|
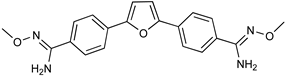 | 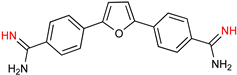 |
| Parafuramidine (DB289) | Furamidine (DB75) |
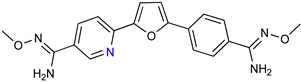 | 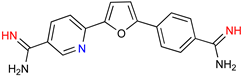 |
| DB844 | DB20 |
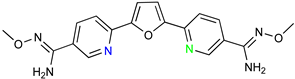 | 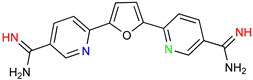 |
| DB868 | DB829 |
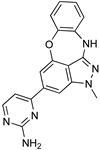
| Recent Pharmaceutical Discovery for the Treatment of HAT and AAT | ||
|---|---|---|
 |  | 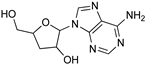 |
| Acoziborole (SCYX-7158) | Tubercidin | Cordycepin |
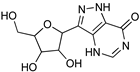 | 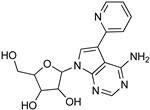 | 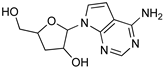 |
| Formycin B | 1 | 2 |
 |  |  |
| 3 | 4 | 5 |
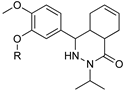 | 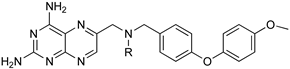 | |
| Diaryl ether substituted tetrahydrophthalazinones scaffold | Pteridine derivatives scaffold | |
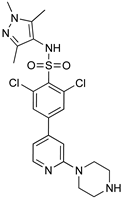 | 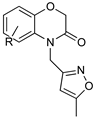 | |
| DDD85646 | Benzomorpholinone scaffold | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturelli, A.; Tagliazucchi, L.; Lima, C.; Venuti, F.; Malpezzi, G.; Magoulas, G.E.; Santarem, N.; Calogeropoulou, T.; Cordeiro-da-Silva, A.; Costi, M.P. Current Treatments to Control African Trypanosomiasis and One Health Perspective. Microorganisms 2022, 10, 1298. https://doi.org/10.3390/microorganisms10071298
Venturelli A, Tagliazucchi L, Lima C, Venuti F, Malpezzi G, Magoulas GE, Santarem N, Calogeropoulou T, Cordeiro-da-Silva A, Costi MP. Current Treatments to Control African Trypanosomiasis and One Health Perspective. Microorganisms. 2022; 10(7):1298. https://doi.org/10.3390/microorganisms10071298
Chicago/Turabian StyleVenturelli, Alberto, Lorenzo Tagliazucchi, Clara Lima, Federica Venuti, Giulia Malpezzi, George E. Magoulas, Nuno Santarem, Theodora Calogeropoulou, Anabela Cordeiro-da-Silva, and Maria Paola Costi. 2022. "Current Treatments to Control African Trypanosomiasis and One Health Perspective" Microorganisms 10, no. 7: 1298. https://doi.org/10.3390/microorganisms10071298