
Use of Whole-Genome Sequencing to Explore Mycobacterium tuberculosis Complex Circulating in a Hotspot Department in France

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Study Population
2.2. Culture and Identification of MTBC
2.3. Phenotypic Drug-Susceptibility Testing
2.4. Whole-Genome Sequencing
2.5. Variant Calling
2.6. Typing and Genotypic Drug Resistance Prediction
2.7. Phylogenetic Analyses and Result Visualization
2.8. Accession Number(s)
2.9. Statistical Analysis
3. Results
3.1. Characteristics of the Strains
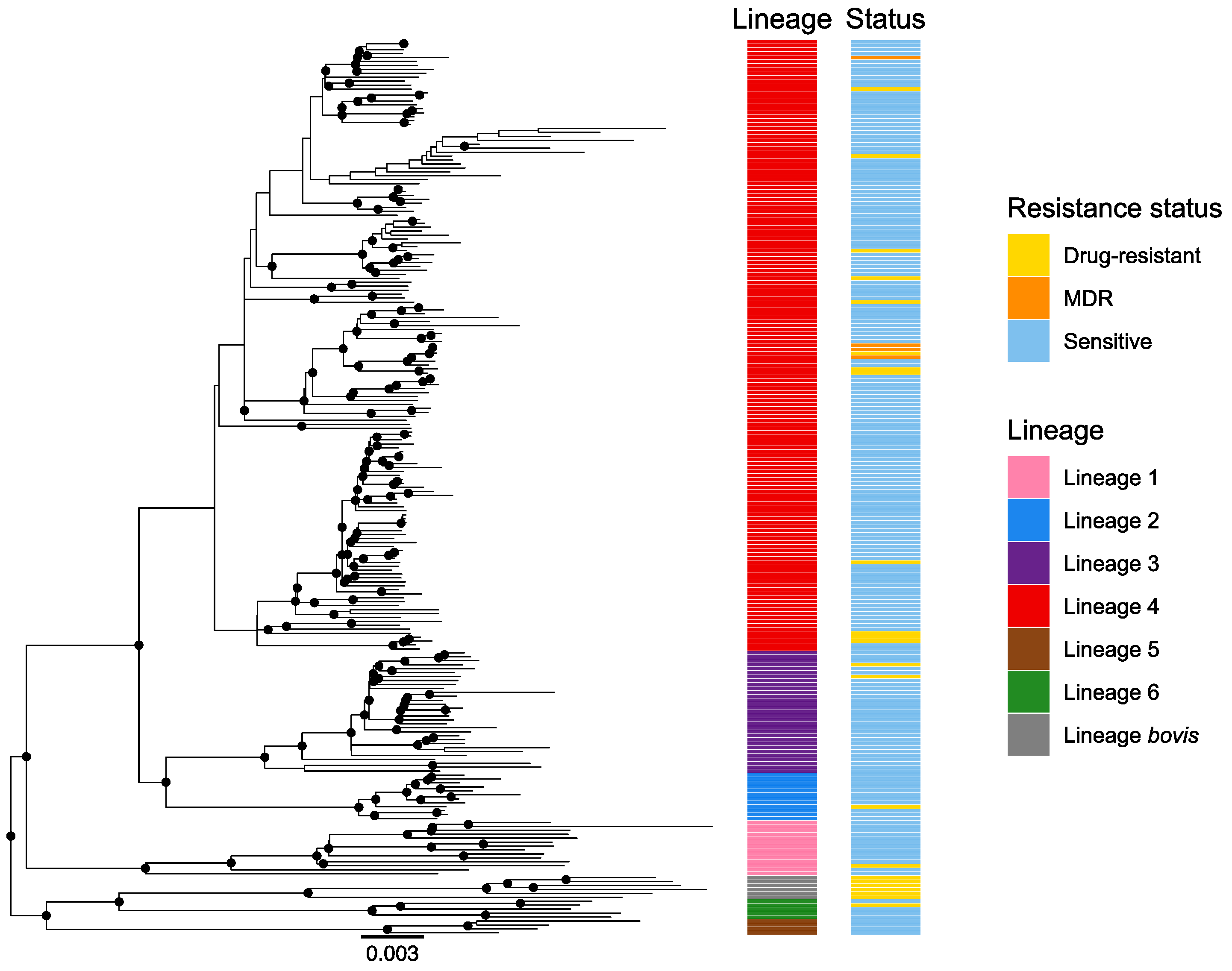
3.2. Lineages Identification and Phylogenetic Analysis
3.3. Drug-Susceptibility Testing
3.4. Comparison of WGS and Phenotypic DST Results
4. Discussion
4.1. Local Epidemiology and Lineage Diversity
4.2. Genomic Versus Phenotypic Approach for Diagnosing Drug Susceptibility and Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2020.
- Cole, S.T.; Brosch, R.; Parkhill, J.A.; Garnier, T.; Churcher, C.; Harris, D.R.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Papaventsis, D.; Casali, N.; Kontsevaya, I.; Drobniewski, F.; Cirillo, D.; Nikolayevskyy, V. Whole genome sequencing of Mycobacterium tuberculosis for detection of drug resistance: A systematic review. Clin. Microbiol. Infect. 2017, 23, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, F.; McNerney, R.; Preston, M.D.; Guerra-Assunção, J.A.; Warry, A.; Hill-Cawthorne, G.; Mallard, K.; Nair, M.; Miranda, A.; Alves, A.; et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Corrales, L.; Tovar-Aguirre, O.L.; Galeano-Vanegas, N.F.; Jiménez, P.A.C.; Martínez-Vega, R.A.; Maldonado-Londoño, C.E.; Hernández-Botero, J.S.; Siller-López, F. Phylogenomic analysis and Mycobacterium tuberculosis antibiotic resistance prediction by whole-genome sequencing from clinical isolates of Caldas, Colombia. PLoS ONE 2021, 16, e0258402. [Google Scholar] [CrossRef]
- Gagneux, S.; DeRiemer, K.; Van, T.; Kato-Maeda, M.; de Jong, B.C.; Narayanan, S.; Nicol, M.; Niemann, S.; Kremer, K.; Gutierrez, M.C.; et al. Variable host–pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2869–2873. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, A.E.; Tsolaki, A.G.; DeRiemer, K.; Feldman, M.W.; Small, P.M. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci. USA 2004, 101, 4871–4876. [Google Scholar] [CrossRef] [Green Version]
- Tessema, B.; Beer, J.; Merker, M.; Emmrich, F.; Sack, U.; Rodloff, A.C.; Niemann, S. Molecular epidemiology and transmission dynamics of Mycobacterium tuberculosis in Northwest Ethiopia: New phylogenetic lineages found in Northwest Ethiopia. BMC Infect. Dis. 2013, 13, 131. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; He, L.; Huang, H.; Shi, C.; Ni, X.; Dai, G.; Ma, L.; Li, W. Mycobacterium tuberculosis Lineage Distribution in Xinjiang and Gansu Provinces, China. Sci. Rep. 2017, 7, 1068. [Google Scholar] [CrossRef] [Green Version]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Ogarkov, O.; Mokrousov, I.; Sinkov, V.; Zhdanova, S.; Antipina, S.; Savilov, E. ‘Lethal’ combination of Mycobacterium tuberculosis Beijing genotype and human CD209 −336G allele in Russian male population. Infect. Genet. Evol. 2012, 12, 732–736. [Google Scholar] [CrossRef]
- Newton, S.M.; Smith, R.J.; Wilkinson, K.A.; Nicol, M.P.; Garton, N.J.; Staples, K.J.; Stewart, G.R.; Wain, J.R.; Martineau, A.R.; Fandrich, S.; et al. A deletion defining a common Asian lineage of Mycobacterium tuberculosis associates with immune subversion. Proc. Natl. Acad. Sci. USA 2006, 103, 15594–15598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthmann, J.-P.; Aït Belghiti, F.; Lévy-Bruhl, D. Epidemiology of Tuberculosis in France in 2015. Impact of the Suspension of the BCG Vaccination Obligation on Childhood Tuberculosis, 2007–2015. Bull. Epidémiol. Hebd. 2017, 7, 116–126. [Google Scholar]
- Castro, A.; Rolland, C.; Silué, Y.; Mangin, F. Tuberculosis Incidence in 2013–2018: How Is Seine-Saint-Denis (France) Different? Bull. Epidémiol. Hebd. 2020, 10–11, 224–231. [Google Scholar]
- Epidémiologie de la tuberculose en France. Données 2020, Santé Publique France. Available online: https://www.santepubliquefrance.fr/maladies-et-traumatismes/maladies-et-infections-respiratoires/tuberculose/donnees/#tabs (accessed on 2 August 2022).
- Billard-Pomares, T.; Bleibtreu, A.; Walewski, V.; Dziri, S.; Barbat, A.; Zahar, J.-R.; Cruaud, P.; Carbonnelle, E. Proposition of a safe Mycobacterium tuberculosis complex denaturation method that does not compromise the integrity of DNA for whole-genome sequencing. Tuberculosis 2019, 117, 62–64. [Google Scholar] [CrossRef]
- MMartin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- GitHub. Samtools 2021 Bcftools, Version 1.11; GitHub: San Francisco, CA, USA, 2020.
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.B.; Warren, R.; Dippenaar, A.; Parsons, S.D.C.; Beisel, C.; Behr, M.A.; et al. A New Phylogenetic Framework for the Animal-Adapted Mycobacterium tuberculosis Complex. Front. Microbiol. 2018, 9, 2820. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2. WIREs Comp. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Scatterpie: Scatter Pie Plot; R Package Version 0.1. 2018. Available online: https://CRAN.R-project.org/package=scatterpie (accessed on 2 August 2022).
- Pierre-Audigier, C.; Talla, C.; Alame-Emane, A.-K.; Audigier, B.; Grall, N.; Ruimy, R.; Andremont, A.; Cadet-Daniel, V.; Sola, C.; Takiff, H.; et al. Tuberculosis trends in a hot-spot region in Paris, France. Int. J. Tuberc. Lung Dis. 2020, 24, 428–435. [Google Scholar] [CrossRef]
- Guthmann, J.-P.; Laporal, S.; Lévy-Bruhl, D. Tuberculosis in France in 2018: Low National Incidence, High Incidence In Certain Geographical Areas and Population Groups. Bull. Epidémiol. Hebd. 2019, 10–11, 196–203. [Google Scholar]
- Reed, M.B.; Pichler, V.K.; McIntosh, F.; Mattia, A.; Fallow, A.; Masala, S.; Domenech, P.; Zwerling, A.; Thibert, L.; Menzies, D.; et al. Major Mycobacterium tuberculosis Lineages Associate with Patient Country of Origin. J. Clin. Microbiol. 2009, 47, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.T.; Gardiner, S.; Smith, E.G.; Webber, R.; Hawkey, P.M. Global Origin of Mycobacterium tuberculosisin the Midlands, UK. Emerg. Infect. Dis. 2010, 16, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Click, E.S.; Moonan, P.; Winston, C.A.; Cowan, L.S.; Oeltmann, J.E. Relationship Between Mycobacterium tuberculosis Phylogenetic Lineage and Clinical Site of Tuberculosis. Clin. Infect. Dis. 2011, 54, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, J.W.; Gillespie, S.H.; McHugh, T.D.; Pitman, R.; Bothamley, G.H.; Drobniewski, F. Origins and properties of Mycobacterium tuberculosis isolates in London. J. Med. Microbiol. 2005, 54, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lari, N.; Rindi, L.; Cristofani, R.; Rastogi, N.; Tortoli, E.; Garzelli, C. Association of Mycobacterium tuberculosis complex isolates of BOVIS and Central Asian (CAS) genotypic lineages with extrapulmonary disease. Clin. Microbiol. Infect. 2009, 15, 538–543. [Google Scholar] [CrossRef]
- Brown, A.C.; Bryant, J.M.; Einer-Jensen, K.; Holdstock, J.; Houniet, D.T.; Chan, J.Z.M.; Depledge, D.P.; Nikolayevskyy, V.; Broda, A.; Stone, M.J.; et al. Rapid Whole-Genome Sequencing of Mycobacterium tuberculosis Isolates Directly from Clinical Samples. J. Clin. Microbiol. 2015, 53, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Khandkar, C.; Harrington, Z.; Jelfs, P.J.; Sintchenko, V.; Dobler, C.C. Epidemiology of Peripheral Lymph Node Tuberculosis and Genotyping of M. tuberculosis Strains: A Case-Control Study. PLoS ONE 2015, 10, e0132400. [Google Scholar] [CrossRef]
- Sankar, M.M.; Singh, J.; Diana, S.C.A.; Singh, S. Molecular characterization of Mycobacterium tuberculosis isolates from North Indian patients with extrapulmonary tuberculosis. Tuberculosis 2012, 93, 75–83. [Google Scholar] [CrossRef]
- Genestet, C.; Tatai, C.; Berland, J.-L.; Claude, J.-B.; Westeel, E.; Hodille, E.; Fredenucci, I.; Rasigade, J.-P.; Ponsoda, M.; Jacomo, V.; et al. Prospective Whole-Genome Sequencing in Tuberculosis Outbreak Investigation, France, 2017–2018. Emerg. Infect. Dis. 2019, 25, 589–592. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.M.; Monk, P.; Smith, E.G.; Peto, T.E.A. Contact investigations for outbreaks of Mycobacterium tuberculosis: Advances through whole genome sequencing. Clin. Microbiol. Infect. 2013, 19, 796–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabibbe, A.M.; Walker, T.M.; Niemann, S.; Cirillo, D.M. Whole genome sequencing of Mycobacterium tuberculosis. Eur. Respir. J. 2018, 52, 1801163. [Google Scholar] [CrossRef] [PubMed]
- Jarlier, V. Rapport D’activité Pour L’année 2018; Centre National de Référence des Mycobactéries et de La Résistance des Mycobactéries Aux Antituberculeux (CNR-MyRMA): Paris, France, 2018. [Google Scholar]
- Genestet, C.; Hodille, E.; Berland, J.-L.; Ginevra, C.; Bryant, J.E.; Ader, F.; Lina, G.; Dumitrescu, O. Whole-genome sequencing in drug susceptibility testing of Mycobacterium tuberculosis in routine practice in Lyon, France. Int. J. Antimicrob. Agents 2020, 55, 105912. [Google Scholar] [CrossRef]
- CChen, X.; He, G.; Wang, S.; Lin, S.; Chen, J.; Zhang, W. Evaluation of Whole-Genome Sequence Method to Diagnose Resistance of 13 Anti-tuberculosis Drugs and Characterize Resistance Genes in Clinical Multi-Drug Resistance Mycobacterium tuberculosis Isolates From China. Front. Microbiol. 2019, 10, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Yuan, W.; Molaeipour, L.; Azizian, K.; Ahmadi, A.; Kouhsari, E. Antibiotic heteroresistance in Mycobacterium tuberculosis isolates: A systematic review and meta-analysis. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 73. [Google Scholar] [CrossRef]
- McIvor, A.; Koornhof, H.; Kana, B.D. Relapse, re-infection and mixed infections in tuberculosis disease. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Huang, F.; Yin, X.; Gu, D. Assessments of Different Methods for Testing Heteroresistance to Rifampicin in Tubercle Bacillus. J. Nanosci. Nanotechnol. 2018, 18, 8414–8418. [Google Scholar] [CrossRef]
- Maningi, N.; Daum, L.T.; Rodriguez, J.D.; Mphahlele, M.; Peters, R.P.H.; Fischer, G.W.; Chambers, J.P.; Fourie, P.B. Improved Detection by Next-Generation Sequencing of Pyrazinamide Resistance in Mycobacterium tuberculosis Isolates. J. Clin. Microbiol. 2015, 53, 3779–3783. [Google Scholar] [CrossRef] [Green Version]
- Chedore, P.; Bertucci, L.; Wolfe, J.; Sharma, M.; Jamieson, F. Potential for Erroneous Results Indicating Resistance When Using the Bactec MGIT 960 System for Testing Susceptibility of Mycobacterium tuberculosis to Pyrazinamide. J. Clin. Microbiol. 2010, 48, 300–301. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, Z.; Permar, S. Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J. Med. Microbiol. 2002, 51, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar] [PubMed]
- Hoffner, S.; Ängeby, K.; Sturegård, E.; Jönsson, B.; Johansson, A.; Sellin, M.; Werngren, J. Proficiency of drug susceptibility testing of Mycobacterium tuberculosis against pyrazinamide: The Swedish experience. Int. J. Tuberc. Lung Dis. 2013, 17, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Piersimoni, C.; Mustazzolu, A.; Giannoni, F.; Bornigia, S.; Gherardi, G.; Fattorini, L. Prevention of False Resistance Results Obtained in Testing the Susceptibility of Mycobacterium tuberculosis to Pyrazinamide with the Bactec MGIT 960 System Using a Reduced Inoculum. J. Clin. Microbiol. 2013, 51, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werngren, J.; Alm, E.; Mansjö, M. Non- pncA Gene-Mutated but Pyrazinamide-Resistant Mycobacterium tuberculosis: Why Is That? J. Clin. Microbiol. 2017, 55, 1920–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Morlock, G.P.; Sikes, R.D.; Dalton, T.L.; Metchock, B.; Starks, A.M.; Hooks, D.P.; Cowan, L.S.; Plikaytis, B.B.; Posey, J.E. Molecular Detection of Mutations Associated with First- and Second-Line Drug Resistance Compared with Conventional Drug Susceptibility Testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2032–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engström, A.; Morcillo, N.; Imperiale, B.; Hoffner, S.E.; Juréen, P. Detection of First- and Second-Line Drug Resistance in Mycobacterium tuberculosis Clinical Isolates by Pyrosequencing. J. Clin. Microbiol. 2012, 50, 2026–2033. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Huang, F.; Zhang, G.; He, W.; Ou, X.; He, P.; Zhao, B.; Zhu, B.; Liu, F.; Li, Z.; et al. Whole-genome sequencing for surveillance of tuberculosis drug resistance and determination of resistance level in China. Clin. Microbiol. Infect. 2021, 28, 731.e9–731.e15. [Google Scholar] [CrossRef]
- Shea, J.; Halse, T.A.; Lapierre, P.; Shudt, M.; Kohlerschmidt, D.; Van Roey, P.; Limberger, R.; Taylor, J.; Escuyer, V.; Musser, K.A. Comprehensive Whole-Genome Sequencing and Reporting of Drug Resistance Profiles on Clinical Cases of Mycobacterium tuberculosis in New York State. J. Clin. Microbiol. 2017, 55, 1871–1882. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.S.; Pai, M. Real-Time Sequencing of Mycobacterium tuberculosis: Are We There Yet? J. Clin. Microbiol. 2017, 55, 1249–1254. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Drug | Phenotypically Resistant | Phenotypically Susceptible | Consistency (%) | WGS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genetically Resistant | Genetically Susceptible | Genetically Susceptible | Genetically Resistant | Undetected Variants | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | ||||||
| RIF | 5 | 0 | 222 | 0 | NA | 100 | 100 | 100 | 100 | 100 | ||||
| INH | 18 | 0 | 209 | 0 | NA | 100 | 100 | 100 | 100 | 100 | ||||
| EMB | 1 | 0 | 224 | 2 | embB A313V (one strain) embB G406A (one strain) | 99.1 | 100 | 99.1 | 33.3 | 100 | ||||
| PZA | 8 | 4 | 215 | 0 | NA | 98.2 | 66.7 | 100 | 100 | 98.2 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billard-Pomares, T.; Marin, J.; Quagliaro, P.; Méchaï, F.; Walewski, V.; Dziri, S.; Carbonnelle, E. Use of Whole-Genome Sequencing to Explore Mycobacterium tuberculosis Complex Circulating in a Hotspot Department in France. Microorganisms 2022, 10, 1586. https://doi.org/10.3390/microorganisms10081586
Billard-Pomares T, Marin J, Quagliaro P, Méchaï F, Walewski V, Dziri S, Carbonnelle E. Use of Whole-Genome Sequencing to Explore Mycobacterium tuberculosis Complex Circulating in a Hotspot Department in France. Microorganisms. 2022; 10(8):1586. https://doi.org/10.3390/microorganisms10081586
Chicago/Turabian StyleBillard-Pomares, Typhaine, Julie Marin, Pauline Quagliaro, Frédéric Méchaï, Violaine Walewski, Samira Dziri, and Etienne Carbonnelle. 2022. "Use of Whole-Genome Sequencing to Explore Mycobacterium tuberculosis Complex Circulating in a Hotspot Department in France" Microorganisms 10, no. 8: 1586. https://doi.org/10.3390/microorganisms10081586
APA StyleBillard-Pomares, T., Marin, J., Quagliaro, P., Méchaï, F., Walewski, V., Dziri, S., & Carbonnelle, E. (2022). Use of Whole-Genome Sequencing to Explore Mycobacterium tuberculosis Complex Circulating in a Hotspot Department in France. Microorganisms, 10(8), 1586. https://doi.org/10.3390/microorganisms10081586