
Regulation of Host Immune Response against Enterobacter cloacae Proteins via Computational mRNA Vaccine Design through Transcriptional Modification

 , , , , , , ,
, , , , , , ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval of Bacterial Protein
2.2. Immunogenic-Antigenic and Allergenic Prediction of Protein
2.3. Immunoinformatics Analysis
2.3.1. CLT Epitopes Prediction
2.3.2. Prediction of B-Cells Epitopes
2.3.3. Prediction of HLT-Epitope
2.3.4. Human Homology
2.3.5. Antigenicity, Allergenicity and Toxicity Assessment of Epitopes
2.3.6. Sequences Alignment
2.3.7. T-Lymphocytes and their MHC-Alleles Molecular Docking Analysis
2.3.8. Population Coverage by IEDB
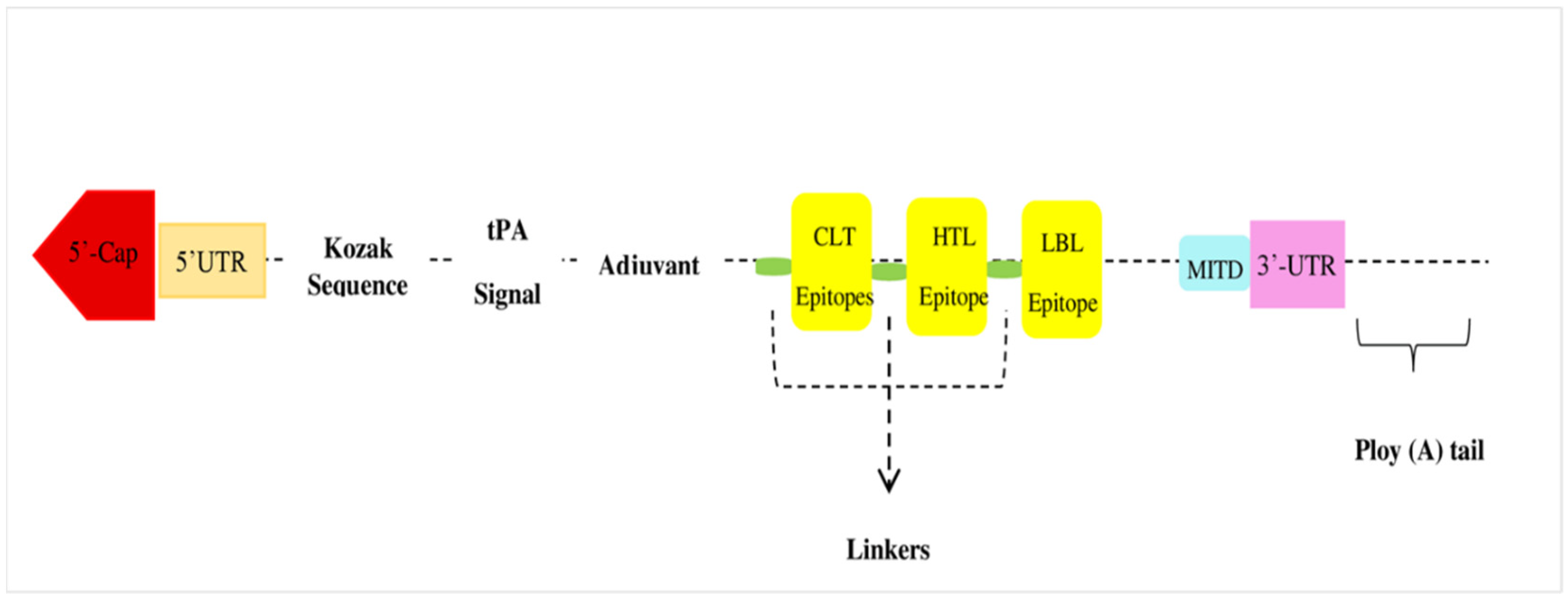
2.4. Vaccine Designing
2.4.1. Evaluation of Physiochemical Profiling of Translated Vaccine
2.4.2. Immune Simulation Response
2.4.3. Optimization of Codon of mRNA Vaccine
2.4.4. Secondary Structure Prediction for mRNA Vaccine
2.4.5. Estimation and Validation of Peptides Structure
2.4.6. Conformation of B-Cells Epitope Prediction
2.4.7. Molecular Docking of Vaccine Construct
2.4.8. Molecular Dynamic Simulation
3. Results
3.1. B-Cell Epitopes Prediction and Evaluation
3.2. CTL Epitopes Prediction and Evaluation
3.3. HTL Epitopes Prediction and Evaluation
3.4. Molecular Docking Interaction of Epitopes and MHC-Alleles
3.5. Vaccine Construct
3.6. Assessment of Physiochemical Profiling of Vaccine Design
3.7. Population Coverage Prediction
3.8. Immune Simulation Response
3.9. Codon Optimization of Construct
3.10. mRNA Vaccine’s Secondary Structure Prediction
3.11. Peptides Structure Prediction of mRNA Vaccine
3.12. Conformational Prediction of B-Cell
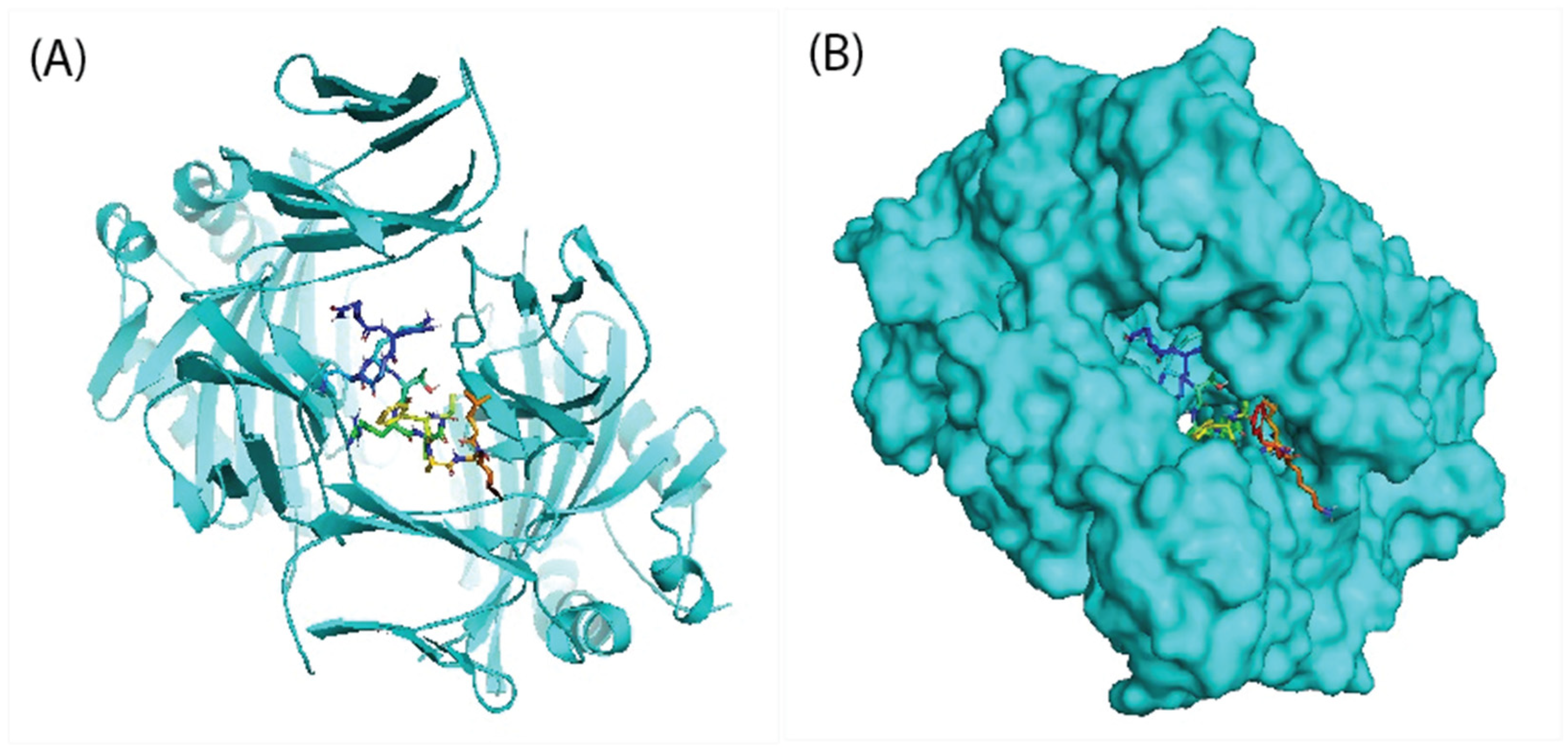
3.13. Molecular Docking of Vaccine Peptides
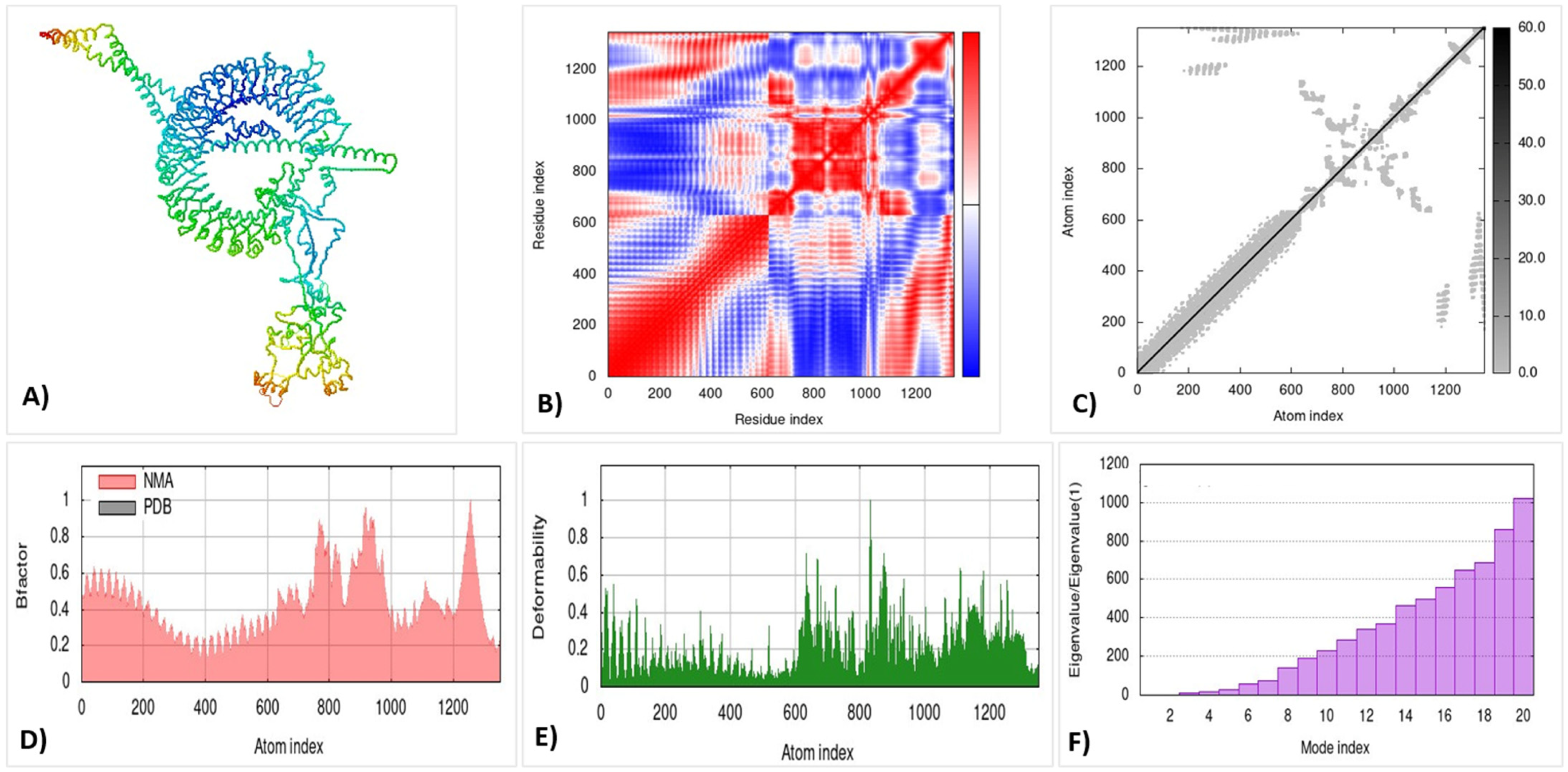
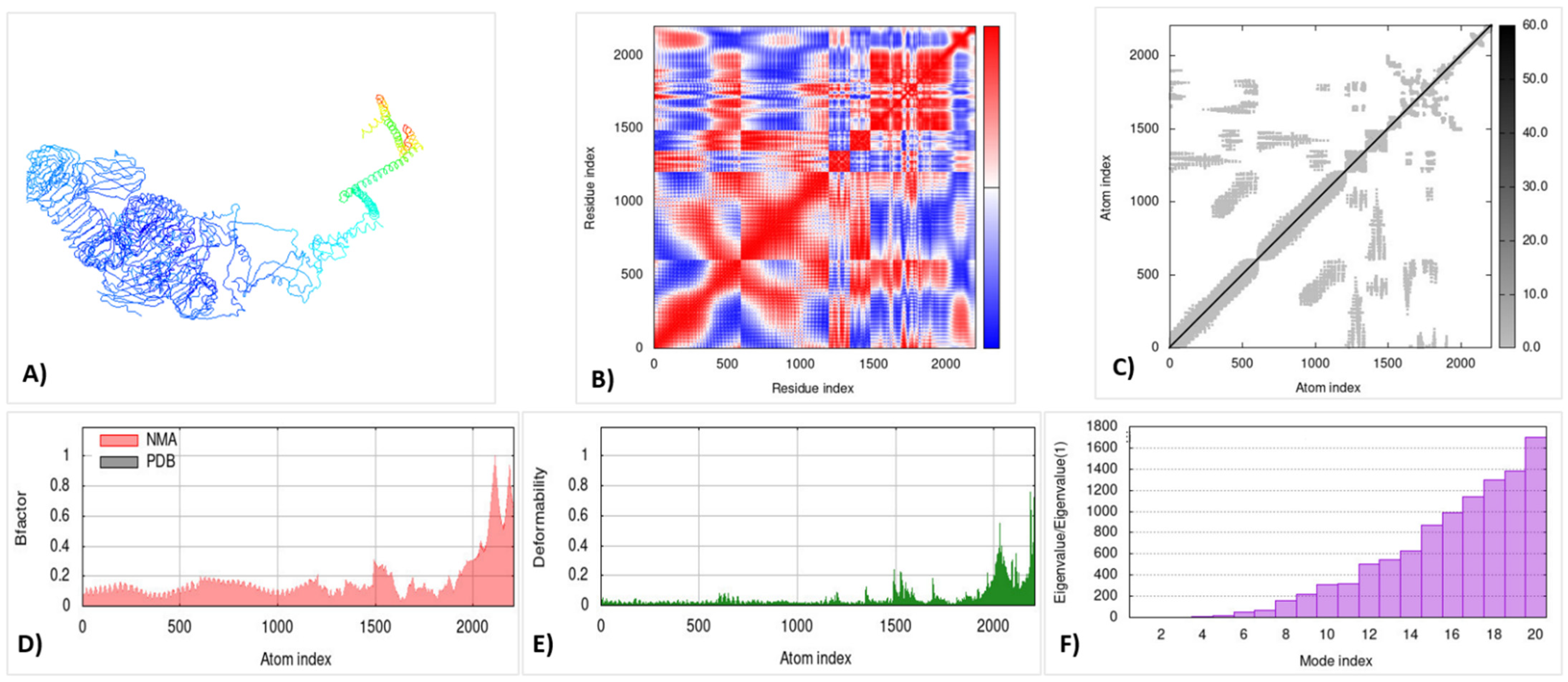
3.14. Molecular Dynamic Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ioannou, P.; Vamvoukaki, R.; Kofteridis, D.P. Infective endocarditis by Enterobacter cloacae: A systematic review and meta-analysis. J. Chemother. 2022, 34, 1–8. [Google Scholar] [CrossRef]
- Zong, X.; Cheng, Y.; Xiao, X.; Fu, J.; Wang, F.; Lu, Z.; Wang, Y.; Jin, M. Protective effects of sulfated polysaccharide from Enterobacter cloacae Z0206 against DSS-induced intestinal injury via DNA methylation. Int. J. Biol. Macromol. 2021, 183, 861–869. [Google Scholar] [CrossRef]
- Davin-Regli, A.; Pagès, J.-M. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front. Microbiol. 2015, 6, 392. [Google Scholar] [CrossRef] [Green Version]
- Rabaan, A.A.; Alhumaid, S.; Mutair, A.A.; Garout, M.; Abulhamayel, Y.; Halwani, M.A.; Alestad, J.H.; Bshabshe, A.A.; Sulaiman, T.; AlFonaisan, M.K. Application of Artificial Intelligence in Combating High Antimicrobial Resistance Rates. Antibiotics 2022, 11, 784. [Google Scholar] [CrossRef]
- Zahra, N.; Zeshan, B.; Qadri, M.M.A.; Ishaq, M.; Afzal, M.; Ahmed, N. Phenotypic and genotypic evaluation of antibiotic resistance of Acinetobacter baumannii bacteria isolated from surgical intensive care unit patients in Pakistan. Jundishapur J. Microbiol. 2021, 14, e113008. [Google Scholar] [CrossRef]
- Cairns, K.A.; Hall, V.; Martin, G.E.; Griffin, D.W.; Stewart, J.D.; Khan, S.F.; Abbott, I.J.; Meher-Homji, Z.; Morrissey, C.O.; Sia, C.; et al. Treatment of invasive IMP-4 Enterobacter cloacae infection in transplant recipients using ceftazidime/avibactam with aztreonam: A case series and literature review. Transpl. Infect. Dis. 2021, 23, e13510. [Google Scholar] [CrossRef]
- Ahmed, N.; Khalid, H.; Mushtaq, M.; Basha, S.; Rabaan, A.A.; Garout, M.; Halwani, M.A.; Al Mutair, A.; Alhumaid, S.; Al Alawi, Z.; et al. The Molecular Characterization of Virulence Determinants and Antibiotic Resistance Patterns in Human Bacterial Uropathogens. Antibiotics 2022, 11, 516. [Google Scholar] [CrossRef]
- Zeb, S.; Mushtaq, M.; Ahmad, M.; Saleem, W.; Rabaan, A.A.; Naqvi, B.S.Z.; Garout, M.; Aljeldah, M.; Al Shammari, B.R.; Al Faraj, N.J.; et al. Self-Medication as an Important Risk Factor for Antibiotic Resistance: A Multi-Institutional Survey among Students. Antibiotics 2022, 11, 842. [Google Scholar] [CrossRef]
- Al Tbeishat, H. Novel In Silico mRNA vaccine design exploiting proteins of M. tuberculosis that modulates host immune responses by inducing epigenetic modifications. Sci. Rep. 2022, 12, 4645. [Google Scholar] [CrossRef]
- Ghosh, D.; Veeraraghavan, B.; Elangovan, R.; Vivekanandan, P. Antibiotic Resistance and Epigenetics: More to It than Meets the Eye. Antimicrob. Agents Chemother. 2020, 64, e02225-19. [Google Scholar] [CrossRef]
- Asif Rasheed, M.; Awais, M.; Aldhahrani, A.; Althobaiti, F.; Alhazmi, A.; Sattar, S.; Afzal, U.; Ali Baeshen, H.; Ali El Enshasy, H.; Joe Dailin, D.; et al. Designing a highly immunogenic multi epitope based subunit vaccine against Bacillus cereus. Saudi J. Biol. Sci. 2021, 28, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, G.F.; Ramesh, K.; Padmanabhan, P.; Gopalan, V.; Govindan, K.; Chandran, A.; Srinivasan, S.; Krishnasamy, K. Immunoinformatic Approach for the identification of T Cell and B Cell Epitopes in the Surface Glycoprotein and Designing a Potent Multiepitope Vaccine Construct Against SARS-CoV-2 including the new UK variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sami, S.A.; Marma, K.K.S.; Mahmud, S.; Khan, M.A.N.; Albogami, S.; El-Shehawi, A.M.; Rakib, A.; Chakraborty, A.; Mohiuddin, M.; Dhama, K.; et al. Designing of a Multi-epitope Vaccine against the Structural Proteins of Marburg Virus Exploiting the Immunoinformatics Approach. ACS Omega 2021, 6, 32043–32071. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.T.; Singh, G.; Ahmed, N.; Saiara, S.A.; Timofeev, V.I.; Ahsan Faruqui, N.; Sharika Ahsan, S.; Tabassum, A.; Nebir, S.S.; Andalib, K.M.S.; et al. Computational designing of a novel subunit vaccine for human cytomegalovirus by employing the immunoinformatics framework. J. Biomol. Struct. Dyn. 2022, 1–23. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Sharma, R.; Grover, A. Immuno-informatics guided designing of a multi-epitope vaccine against Dengue and Zika. J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef]
- Hossan, M.I.; Chowdhury, A.S.; Hossain, M.U.; Khan, M.A.; Mahmood, T.B.; Mizan, S. Immunoinformatics aided-design of novel multi-epitope based peptide vaccine against Hendra henipavirus through proteome exploration. Inform. Med. Unlocked 2021, 25, 100678. [Google Scholar] [CrossRef]
- Kumar, S.; Shuaib, M.; Prajapati, K.S.; Singh, A.K.; Choudhary, P.; Singh, S.; Gupta, S. A candidate triple-negative breast cancer vaccine design by targeting clinically relevant cell surface markers: An integrated immuno and bio-informatics approach. 3 Biotech 2022, 12, 72. [Google Scholar] [CrossRef]
- Asadollahi, P.; Pakzad, I.; Sadeghifard, N.; Ghafourian, S.; Kazemian, H.; Kaviar, V.H.; Fattahi, R.; Kalani, B.S. Immunoinformatics Insights into the Internalin A and B Proteins to Design a Multi-Epitope Subunit Vaccine for L. monocytogenes. Int. J. Pept. Res. Ther. 2022, 28, 47. [Google Scholar] [CrossRef]
- Khan, T.; Suleman, M.; Ali, S.S.; Sarwar, M.F.; Ali, I.; Ali, L.; Khan, A.; Rokhan, B.; Wang, Y.; Zhao, R.; et al. Subtractive proteomics assisted therapeutic targets mining and designing ensemble vaccine against Candida auris for immune response induction. Comput. Biol. Med. 2022, 145, 105462. [Google Scholar] [CrossRef]
- Naveed, M.; Tehreem, S.; Arshad, S.; Bukhari, S.A.; Shabbir, M.A.; Essa, R.; Ali, N.; Zaib, S.; Khan, A.; Al-Harrasi, A.; et al. Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat SARS-CoV-2 strains. J. Infect. Public Health 2021, 14, 938–946. [Google Scholar] [CrossRef]
- Kumar, J.; Qureshi, R.; Sagurthi, S.R.; Qureshi, I.A. Designing of Nucleocapsid Protein Based Novel Multi-epitope Vaccine Against SARS-CoV-2 Using Immunoinformatics Approach. Int. J. Pept. Res. Ther. 2021, 27, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.; Kadivella, M.; Sharma, R.; Faisal, S.M.; Azam, S. Designing of multiepitope-based vaccine against Leptospirosis using Immuno-Informatics approaches. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shahrear, S.; Islam, A. Immunoinformatics guided modeling of CCHF_GN728, an mRNA-based universal vaccine against Crimean-Congo hemorrhagic fever virus. Comput. Biol. Med. 2021, 140, 105098. [Google Scholar] [CrossRef]
- Sharma, D.; Patel, S.; Padh, H.; Desai, P. Immunoinformatic Identification of Potential Epitopes Against Shigellosis. Int. J. Pept. Res. Ther. 2016, 22, 481–495. [Google Scholar] [CrossRef]
- Medha; Priyanka; Sharma, S.; Sharma, M. Design of a peptide-based vaccine from late stage specific immunogenic cross-reactive antigens of PE/PPE proteins of Mycobacterium tuberculosis. Eur. J. Pharm. Sci. 2022, 168, 106051. [Google Scholar] [CrossRef] [PubMed]
- Elhassan, R.M.; Alsony, N.M.; Othman, K.M.; Izz-Aldin, D.T.; Alhaj, T.A.; Ali, A.A.; Abashir, L.A.; Ahmed, O.H.; Hassan, M. Computational vaccinology approach: Designing an efficient multi-epitope peptide vaccine against Cryptococcus neoformans var. grubii’s heat shock 70 KDa protein. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Oluwagbemi, O.O.; Oladipo, E.K.; Dairo, E.O.; Ayeni, A.E.; Irewolede, B.A.; Jimah, E.M.; Oyewole, M.P.; Olawale, B.M.; Adegoke, H.M.; Ogunleye, A.J. Computational construction of a glycoprotein multi-epitope subunit vaccine candidate for old and new South-African SARS-CoV-2 virus strains. Inform. Med. Unlocked 2022, 28, 100845. [Google Scholar] [CrossRef]
- Lefranc, M.-P. Immunoinformatics of the V, C, and G domains: IMGT® definitive system for IG, TR and IgSF, MH, and MhSF. In Immunoinformatics; Springer: Berlin/Heidelberg, Germany, 2014; pp. 59–107. [Google Scholar]
- Nain, Z.; Karim, M.M.; Sen, M.K.; Adhikari, U.K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans-An integrated vaccinomics approach. Mol. Immunol. 2020, 120, 146–163. [Google Scholar] [CrossRef]
- Ashraf, N.M.; Bilal, M.; Mahmood, M.S.; Hussain, A.; Mehboob, M.Z. In-silico analysis of putative HCV epitopes against Pakistani human leukocyte antigen background: An approach towards development of future vaccines for Pakistani population. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 43, 58–66. [Google Scholar] [CrossRef]
- Naveed, M.; Ali, U.; Karobari, M.I.; Ahmed, N.; Mohamed, R.N.; Abullais, S.S.; Kader, M.A.; Marya, A.; Messina, P.; Scardina, G.A. A Vaccine Construction against COVID-19-Associated Mucormycosis Contrived with Immunoinformatics-Based Scavenging of Potential Mucoralean Epitopes. Vaccines 2022, 10, 664. [Google Scholar] [CrossRef]
- Naveed, M.; Bukhari, B.; Afzal, N.; Sadia, H.; Meer, B.; Riaz, T.; Ali, U.; Ahmed, N. Geographical, Molecular, and Computational Analysis of Migraine-Causing Genes. J. Comput. Biophys. Chem. 2021, 20, 391–403. [Google Scholar] [CrossRef]
- Naveed, M.; Yaseen, A.R.; Khalid, H.; Ali, U.; Rabaan, A.A.; Garout, M.; Halwani, M.A.; Al Mutair, A.; Alhumaid, S.; Al Alawi, Z. Execution and Design of an Anti HPIV-1 Vaccine with Multiple Epitopes Triggering Innate and Adaptive Immune Responses: An Immunoinformatic Approach. Vaccines 2022, 10, 869. [Google Scholar] [CrossRef] [PubMed]
- Pavitrakar, D.V.; Atre, N.M.; Tripathy, A.S.; Shil, P. Design of a multi-epitope peptide vaccine candidate against chandipura virus: An immuno-informatics study. J. Biomol. Struct. Dyn. 2022, 40, 648–659. [Google Scholar] [CrossRef]
- Soltan, M.A.; Eldeen, M.A.; Elbassiouny, N.; Mohamed, I.; El-damasy, D.A.; Fayad, E.; Abu Ali, O.A.; Raafat, N.; Eid, R.A.; Al-Karmalawy, A.A. Proteome Based Approach Defines Candidates for Designing a Multitope Vaccine against the Nipah Virus. Int. J. Mol. Sci. 2021, 22, 9330. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein * | UniPort Id ** | A. Score *** |
|---|---|---|
| OmpF | V5ISF4 | 0.6395 |
| OmpD | A0A2T4Y430 | 0.6660 |
| Omp35 | G3LW48 | 0.6882 |
| OmpC | Q93K99 | 0.7882 |
| Cell Type | Sequence of Epitope |
|---|---|
| HTL | QNGNKTRLAFAGLKF |
| VAQYQFDFGLRPSIA | |
| YFNKNMSTYVDYKIN | |
| NKNMSTYVDYKINLL | |
| NIYLASTYSETRNMT | |
| QNGNKTRLAFAGLKF | |
| NGNKTRLAFAGLKFG | |
| YFNKNMSTYVDYKIN | |
| CTL | DNTYARLGFK |
| YGKAVGLHYF | |
| AITSSLAVPV | |
| NTYARLGFK | |
| TGYGQWEYNF | |
| WATSLSYDF | |
| AQYQFDFGL | |
| MSTYVDYQIN | |
| KTYVRLGFK | |
| MSTYVDYKI | |
| SGYGQWEYEF | |
| AQYQGKNNK | |
| GYGQWEYEF | |
| MSTYVDYKI | |
| KVLSLLVPAL | |
| KYVDVGATYY | |
| FGLRPSVAYL | |
| VLSLLVPAL | |
| B Lymphocytes | GLHYFSDNDSNDGDNT |
| AGAANAAEIYNKDGNK | |
| YIDVGATYYFNKNMST | |
| IGDEDYINYIDVGATY | |
| SGYGQWEYEFKGNNDE | |
| AGVVNAAEIYNKDGNK | |
| PEFGGDTYGSDNFMQQ | |
| YGQWEYQIQGNSGENE |
| Protein | CLT Epitopes | MHC-I Binding Alleles | HLT Epitope | MHC-II Binding Alleles |
|---|---|---|---|---|
| OmpF | AQYQFDFGL | HLA-A*02:06 | VAQYQFDFGLRPSIA | HLA-DRB3*01:01, HLA-DRB1*04:05, HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01 |
| KSKAKDVEG | HLA-A*30:01 | QNGNKTRLAFAGLKF | HLA-DRB5*01:01, HLA-DQA1*01:02/DQB1*06:02 | |
| WATSLSYDF | HLA-B*35:01, HLA-B*53:01 | |||
| MSTYVDYQIN | HLA-B*58:01, HLA-A*68:02 | |||
| OmpD | AQYQGKNNK | HLA-A*11:01 | YFNKNMSTYVDYKIN | HLA-DRB1*15:01, HLA-DRB1*07:01, HLA-DRB1*13:02, HLA-DRB1*09:01, HLA-DRB3*01:01, HLA-DRB1*01:01, HLA-DRB1*04:05 |
| GYGQWEYEF | HLA-A*23:01 | NIYLASTYSETRNMT | HLA-DRB1*04:05 HLA-DRB1*08:02 HLA-DRB1*04:01 HLA-DRB3*02:02 | |
| KTYVRLGFK | HLA-A*30:01, HLA-A*03:01, HLA-A*11:01, HLA-A*31:01, HLA-A*68:01 | |||
| MSTYVDYKI | HLA-A*68:02, HLA-B*58:01, HLA-B*53:01 | |||
| SGYGQWEYEF | HLA-A*23:01, HLA-A*24:02 | |||
| Omp35 | DNTYARLGFK | HLA-A*11:01, HLA-A*03:01 | QNGNKTRLAFAGLKF | HLA-DRB5*01:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DRB1*09:01, HLA-DRB1*15:01, HLA-DRB1*07:01, HLA-DRB1*01:01 |
| YGKAVGLHYF | HLA-B*15:01, HLA-A*23:01 | NGNKTRLAFAGLKFG | HLA-DPA1*01:03/DPB1*02:01, HLA-DRB5*01:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DRB1*15:01, HLA-DRB1*09:01, HLA-DRB1*07:01, HLA-DRB1*11:01, HLA-DRB1*01:01 | |
| AITSSLAVPV | HLA-A*02:03, HLA-A*02:06, HLA-A*68:02, HLA-A*02:01 | |||
| NTYARLGFK | HLA-A*68:01, HLA-A*03:01, HLA-A*11:01, HLA-A*30:01, HLA-A*33:01, HLA-A*31:01, HLA-A*26:01 | |||
| OmpC | KVLSLLVPAL | HLA-A*02:01, HLA-A*02:06 | YFNKNMSTYVDYKIN | HLA-DRB1*15:01, HLA-DQA1*01:01/DQB1*05:01, HLA-DRB3*02:02, HLA-DRB1*07:01, HLA-DRB1*13:02, HLA-DRB1*09:01, HLA-DRB3*01:01, HLA-DRB1*01:01, HLA-DRB1*04:05 |
| MSTYVDYKI | HLA-A*68:02, HLA-B*58:01, HLA-B*53:01 | NKNMSTYVDYKINLL | HLA-DRB1*15:0, HLA-DRB1*03:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DPA1*01:03/DPB1*02:01 | |
| KYVDVGATYY | HLA-A*01:01, HLA-A*30:02 | |||
| FGLRPSVAYL | HLA-A*02:03, HLA-A*02:01, HLA-B*15:01, HLA-A*02:06 | |||
| VLSLLVPAL | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06 |
| Type of Lymphocytes | Epitopes | Alleles | PDB Id | Binding Affinity (kcal/mol) |
|---|---|---|---|---|
| HTL | YFNKNMSTYVDYKIN | HLA-DRB1*01:01 | 2FSE | −666.9 |
| QNGNKTRLAFAGLKF | HLA-DRB1*15:01 | 1BX2 | −698.6 | |
| CLT | DNTYARLGFK | HLA-A*11:01 | 6ID4 | −562.0 |
| KVLSLLVPAL | HLA-A*02:06 | 3OXR | −544.5 | |
| WATSLSYDF | HLA-B*35:01 | 4PR5 | −569.8 |
| Physiochemical Profiling | Measurement | Indication |
|---|---|---|
| Number of Amino Acid | 717 | Appropriate |
| Number of Atoms | 10,708 | - |
| Molecular Weight | 77,612.39 | Appropriate |
| Formula | C3536H5202N910O1048S12 | - |
| Theoretical pI | 8.75 | Basic |
| Total number of negatively charged residues (Asp + Glu) | 59 | - |
| Total number of positively charged residues (Arg + Lys) | 67 | - |
| Instability Index (II) | 29.53 | Stable |
| Aliphatic index | 60.08 | Thermostable |
| Grand average of hydropathicity (GRAVY) | −0.456 | Hydrophilic |
| Antigenicity (by VaxiJen) | 0.8371 | Antigenic |
| Antigenicity (by ANTIGENpro) | 0.802068 | Antigenic |
| Allergenicity | Non-Allergenic | Non-Allergen |
| Toxicity | Non-Toxic | Non-Toxic |
| Solubility (m/L) | 0.591434 | Soluble |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naveed, M.; Jabeen, K.; Naz, R.; Mughal, M.S.; Rabaan, A.A.; Bakhrebah, M.A.; Alhoshani, F.M.; Aljeldah, M.; Shammari, B.R.A.; Alissa, M.; et al. Regulation of Host Immune Response against Enterobacter cloacae Proteins via Computational mRNA Vaccine Design through Transcriptional Modification. Microorganisms 2022, 10, 1621. https://doi.org/10.3390/microorganisms10081621
Naveed M, Jabeen K, Naz R, Mughal MS, Rabaan AA, Bakhrebah MA, Alhoshani FM, Aljeldah M, Shammari BRA, Alissa M, et al. Regulation of Host Immune Response against Enterobacter cloacae Proteins via Computational mRNA Vaccine Design through Transcriptional Modification. Microorganisms. 2022; 10(8):1621. https://doi.org/10.3390/microorganisms10081621
Chicago/Turabian StyleNaveed, Muhammad, Khizra Jabeen, Rubina Naz, Muhammad Saad Mughal, Ali A. Rabaan, Muhammed A. Bakhrebah, Fahad M. Alhoshani, Mohammed Aljeldah, Basim R. Al Shammari, Mohammed Alissa, and et al. 2022. "Regulation of Host Immune Response against Enterobacter cloacae Proteins via Computational mRNA Vaccine Design through Transcriptional Modification" Microorganisms 10, no. 8: 1621. https://doi.org/10.3390/microorganisms10081621
APA StyleNaveed, M., Jabeen, K., Naz, R., Mughal, M. S., Rabaan, A. A., Bakhrebah, M. A., Alhoshani, F. M., Aljeldah, M., Shammari, B. R. A., Alissa, M., Sabour, A. A., Alaeq, R. A., Alshiekheid, M. A., Garout, M., Almogbel, M. S., Halwani, M. A., Turkistani, S. A., & Ahmed, N. (2022). Regulation of Host Immune Response against Enterobacter cloacae Proteins via Computational mRNA Vaccine Design through Transcriptional Modification. Microorganisms, 10(8), 1621. https://doi.org/10.3390/microorganisms10081621