
A Case Study for the Recovery of Authentic Microbial Ancient DNA from Soil Samples


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
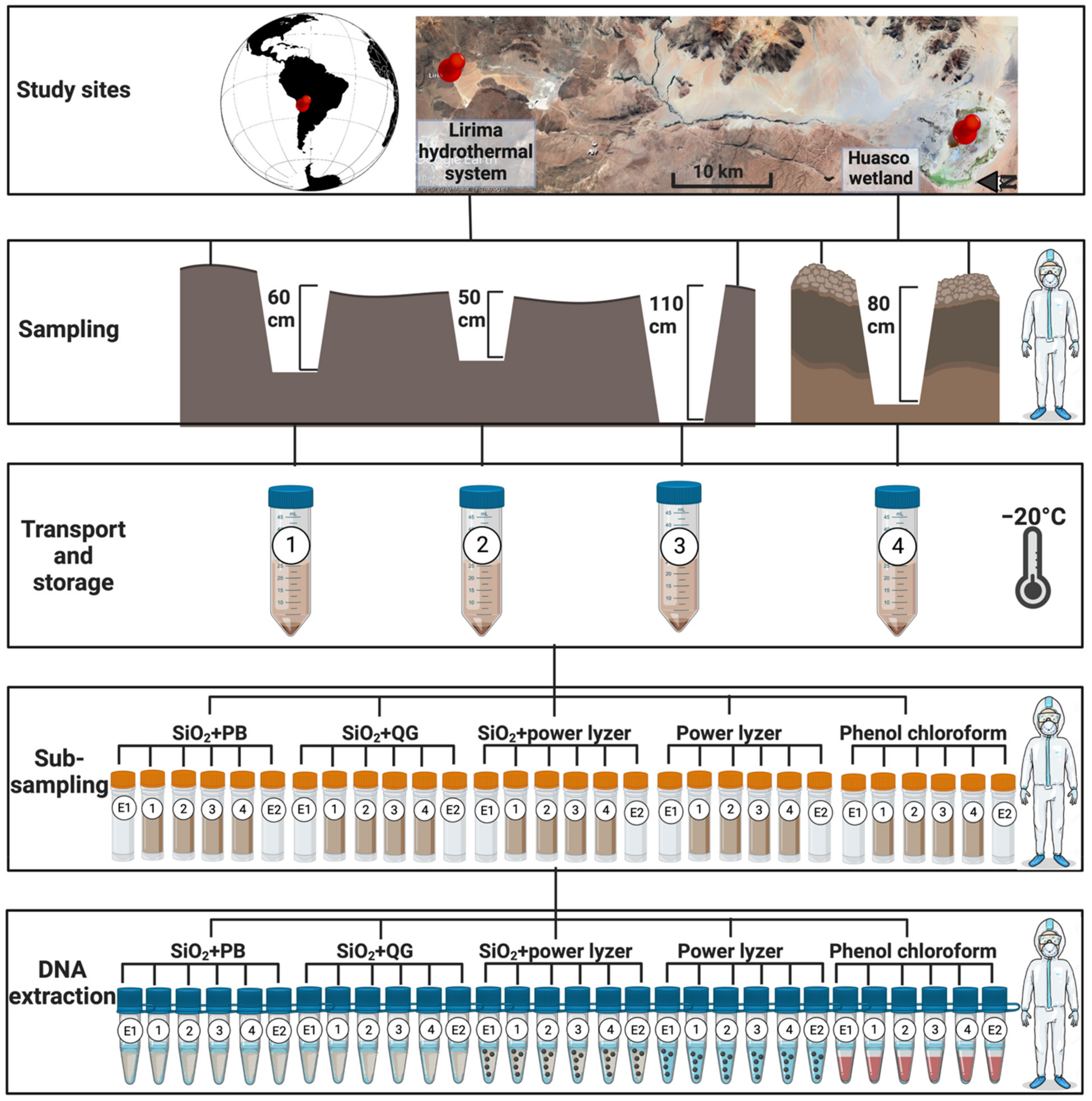
2.1. Sample Location
2.2. Sample Collection and Storage Conditions
2.3. Sample Preparation in aDNA Facilities
2.4. DNA Extraction Methods in aDNA Facilities
- (a)
- SiO2 + PowerLyzer kit (CP): performed using an in-house DNeasy® PowerLyzer® PowerSoil® kit (Qiagen, Hilden, Germany) and silica-based method. We followed PowerLyzer kit protocol using 250 mg of soil samples up to step 10 of the manufacturer’s instructions (solution C3 and centrifugation). We then obtained the supernatant and used an in-house QG buffer (guanidine thiocyanate DNA-binding buffer) and a silica-based method, as previously described [17] with minor modifications. Briefly, we added the supernatant to 3 mL of binding buffer (2.8 mL QG buffer (Qiagen), 46 μL water, 15 μL NaCl (5M), 39 μL Triton-X 100 (Sigma-Aldrich, Saint Louis, MO, USA) and 167 μL acetic acid (3 M)) into a 15 mL tube, added 100 μL of medium-sized silica suspension and mixed under slow and constant rotation. We centrifuged the samples at 4500 rpm for 5 min and the pellet was washed twice by resuspension in 900 μL of 80% ethanol in a 1.5 mL tube. Tubes were centrifuged for 1 min at 14,000 rpm, and the supernatant was removed. The pellet was left to dry at 37 °C for 15 min, subsequently resuspended in 75 μL of pre-warmed (to 50 °C) TLE buffer (10 mM Tris-HCl, 1 mM EDTA), and incubated for 10 min. After pelleting for 1 min at 13,000 rpm, the supernatant was collected, aliquoted, and stored at −20 °C until further use.
- (b)
- SiO2 + PB buffer (PB): performed using a modified PB buffer (guanidine hydrochloride DNA-binding buffer) and silica-based method [34]. Briefly, 250 mg of soil samples was incubated overnight under slow, constant rotation at 37 °C in 1 mL of lysis buffer (900 μL EDTA; 80 μL SDS; 20 μL 20 mg/mL proteinase K). After lysis, samples were centrifuged at 14,500 rpm for 3 min, and the supernatant was added to 12.6 mL of binding buffer (12.2 mL PB buffer (Qiagen), 7 μL Tween-20, and 378 μL acetic acid (3 M)) in a 15 mL tube, with a 100 μL of medium-sized silica suspension and mixed under slow and constant rotation. We centrifuged the samples at 4500 rpm for 5 min, and the pellet was washed twice by resuspension in 900 μL of 80% ethanol in a 1.5 mL tube. Tubes were centrifuged for 1 min at 14,000 rpm, and the supernatant was removed. The pellet was left to dry at 37 °C for 15 min, subsequently resuspended in 75 μL of pre-warmed (to 50 °C) TLE buffer (10 mM Tris-HCl, 1 mM EDTA), and incubated for 10 min. After pelleting for 1 min at 13,000 rpm, the supernatant was collected, aliquoted, and stored at −20 °C until further use.
- (c)
- Phenol–chloroform (PHCH): completed as previously described [35] with minor modifications. Briefly, an equal volume of molecular grade phenol:chloroform was added to 250 mg of soil samples and mixed until an emulsion was formed. The solution was centrifuged at 12,000 rpm for 1 min at room temperature until both phases were separated. The aqueous phase was transferred to a fresh tube, and the process was repeated a second time. Then, an equal volume of chloroform was added, and the solution was mixed and centrifuged at 12,000 rpm for 1 min. The aqueous phase was transferred to a fresh tube. The DNA was precipitated by adding sodium acetate to a final concentration of 0.3 M and mixing the solution. Then, two volumes of ice-cold ethanol and 0.01 M of MgCl2 were added and mixed again. The solution was incubated at −20 °C for 30 min and centrifuged at 14,000 rpm for 15 min at room temperature. The supernatant was removed, and 1 mL of 70% ethanol was added. The solution was centrifuged at 14,000 rpm at 4 °C for 2 min, and the supernatant was removed. The tube was stored open in the hood at room temperature until all the ethanol was evaporated. The DNA pellet was redissolved in 75 μL of TLE buffer.
- (d)
- PowerLyzer kit (PL): DNA extraction of 250 mg of soil sample was performed using the DNeasy® PowerLyzer®PowerSoil® kit (Qiagen) and a Precellys 24 homogenizer (Bertin Instruments, Montigny-le-Bretonneux, France) according to the manufacturer’s instructions.
- (e)
- SiO2 + QG buffer (QG): performed using an in-house QG buffer (guanidine thiocyanate DNA-binding buffer) and silica-based method [17] with minor modifications. Briefly, 250 mg of soil sample were incubated overnight under slow, constant rotation at 37 °C in 1.72 mL lysis buffer (1.6 mL EDTA; 200 uL SDS; 20 uL 20 mg/mL proteinase K). After lysis, samples were centrifuged at 14,500 rpm for 2 min, and the supernatant was added to 3 mL of binding buffer (3.7 mL QG buffer (Qiagen), 61.4 μL water, 20 μL NaCl (5 M) and 52.1 μL Triton-X 100 (Sigma-Aldrich, Saint Louis, MO, USA) in a 15 mL tube, alongside 100 μL of medium-sized silica suspension. The solution was mixed under slow and constant rotation for one hour. We centrifuged the samples at 4500 rpm for 5 min and the pellet was washed two times by resuspension in 900 μL of 80% ethanol in a 1.5 mL tube. Tubes were centrifuged for 1 min at 14,000 rpm and the supernatant was removed. The pellet was left to dry at 37 °C for 15 min, resuspended in 75 μL of pre-warmed (to 50 °C) TLE buffer (10 mM Tris-HCl, 1 mM EDTA), and incubated for 10 min. After pelleting for 1 min at 13,000 rpm, the supernatant was collected, aliquoted, and stored at −20 °C until further use.
2.5. DNA Library Preparation and Shotgun Sequencing
2.6. Data Preprocessing
- (1)
- Pre-filtered pipeline: the raw data were demultiplexed, adapter trimmed and merged using AdapterRemoval v.2.2.1 based on unique P5/P7 barcodes.
- (2)
- Post-filtered 30 kx: the raw data were demultiplexed, adapter trimmed and merged, using AdapterRemoval v.2.2.1. A low-complexity threshold of 30% was then applied using Komplexity v.0.3.6 followed by read deduplication that removed only exact sequences, using the dedupe tool of BBMap v.36.62 (https://sourceforge.net/projects/bbmap/; accessed on 1 December 2020).
- (3)
- Post-filtered 55 kx: the raw data were demultiplexed, adapter trimmed and merge using AdapterRemoval v.2.2.1. A low-complexity threshold of 55% was applied using Komplexity v.0.3.6 followed by read deduplication by removing exact sequences, using the dedupe tool of BBMap v.36.62.
- (4)
- Post-filtered 55 kx_2mm: the raw data were demultiplexed, adapter trimmed and merged using AdapterRemoval v.2.2.1. A low-complexity threshold of 55% was applied using Komplexity v.0.3.6 followed by reads deduplication by removing exact sequences and sequences with two mismatches, using the dedupe tool of BBMap v.36.62.
- (5)
- Post-filtered 70 kx: the raw data were demultiplexed, adapter trimmed and merged using AdapterRemoval v.2.2.1. A low-complexity threshold of 70% was applied using Komplexity v.0.3.6 followed by read deduplication by removing exact sequences, using the dedupe tool of BBMap v.36.62.
- (6)
- Post-filtered fastp_30 kx: the raw data were demultiplexed, adapter trimmed, merged, and a low-complexity threshold of 30% was applied, using Fastp v.0.20.0. Then, collapsed reads were deduplicated by removing exact sequences using the dedup tool of BBMap v.36.62.
- (7)
- Post-filtered fastp_30 kx_2mm: the raw data were demultiplexed, adapter trimmed, and merged, and a low-complexity threshold of 30% was applied, using Fastp v.0.20.0. Then, collapsed reads were deduplicated by removing exact sequences and sequences with two mismatches, using the dedupe tool of BBMap v.36.62.
- (8)
- Post-filtered fastp_55 kx: the raw data were demultiplexed, adapter trimmed, and merged, and a low-complexity threshold of 55% was applied, using Fastp v.0.20.0. Then, collapsed reads were deduplicated by removing exact sequences using the dedup tool of BBMap v.36.62.
2.7. Taxonomic Classification
2.7.1. Impact of DNA Extraction Methods and Bioinformatic Pipelines on Taxonomic Composition
2.7.2. Impact of Databases Selection on Taxonomic Composition
2.8. Authentication of Microbial aDNA
3. Results and Discussion
3.1. How to Collect and Process Soil Samples for Reproductible Environmental aDNA Studies?
3.1.1. Sample Collection
3.1.2. Sample Preparation and Subsampling in aDNA Facilities
3.2. How Are Laboratory Practices and Bioinformatic Workflows Affecting the Reconstruction of Ancient Soil Microbiota?
3.3. Benchmarking Laboratory Methods and Bioinformatic Analysis for the Recovery of Microbial Ancient DNA from Soil Samples
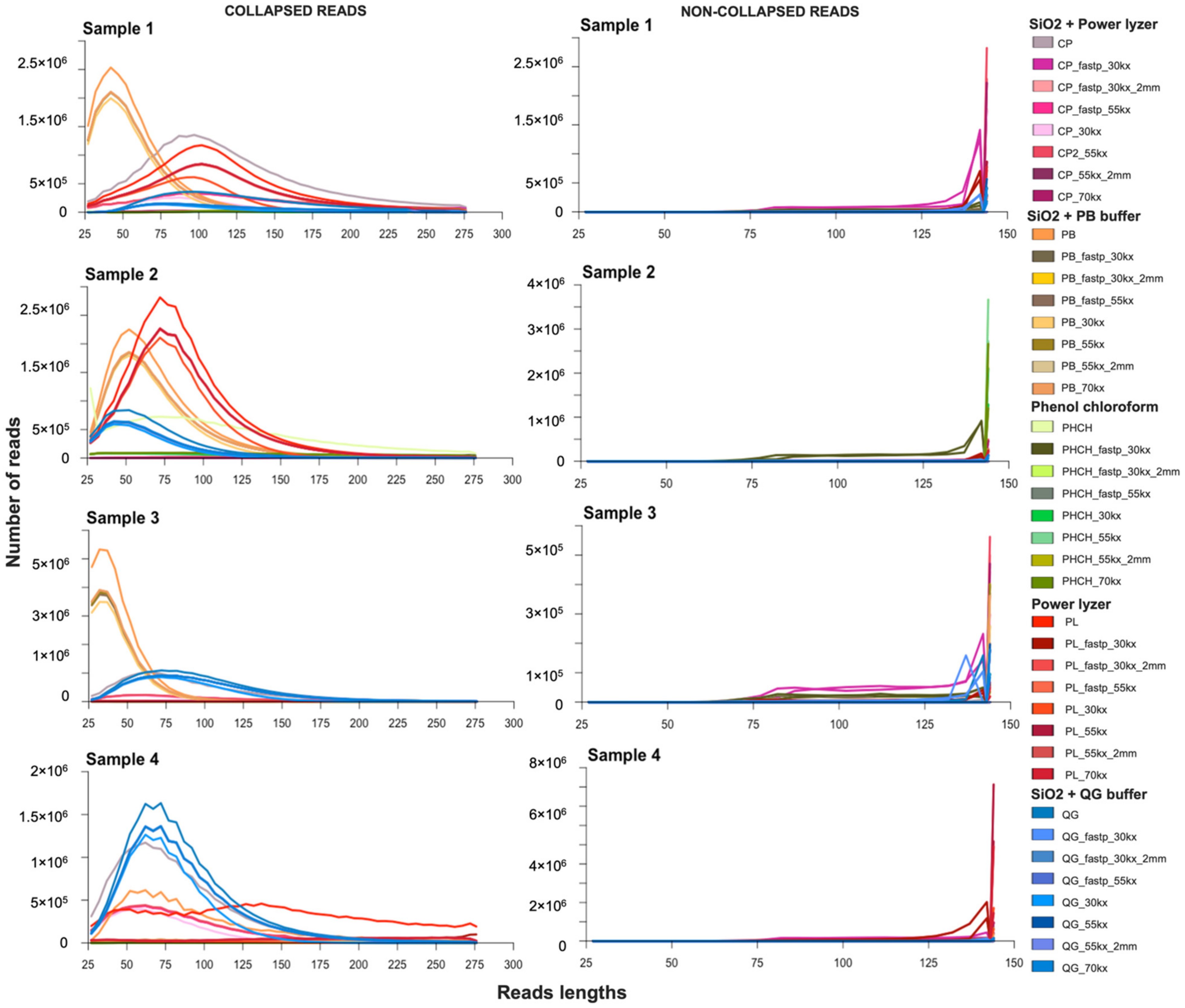
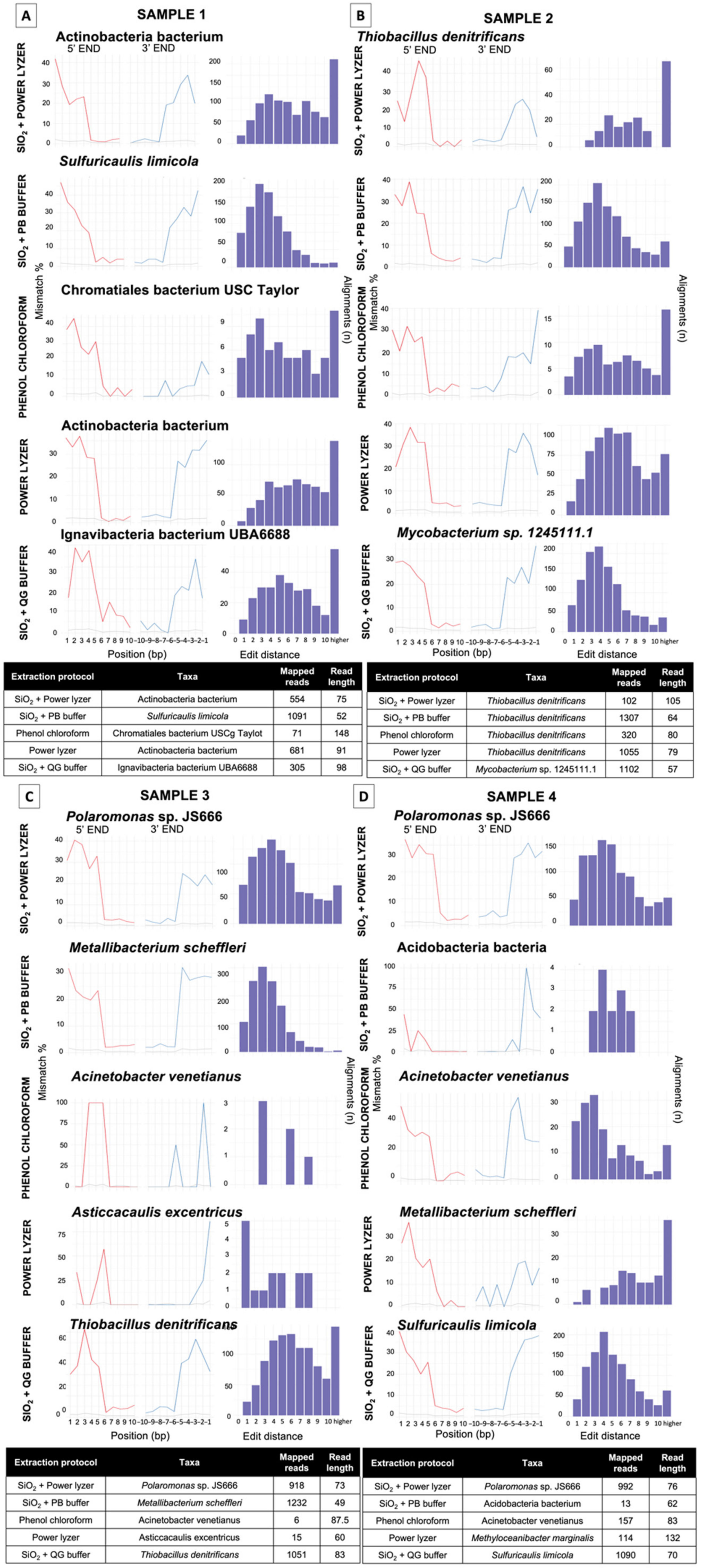
3.3.1. Impact of Extraction Methods and Data Preprocessing Pipelines on Read Number and Fragment Length Recovery
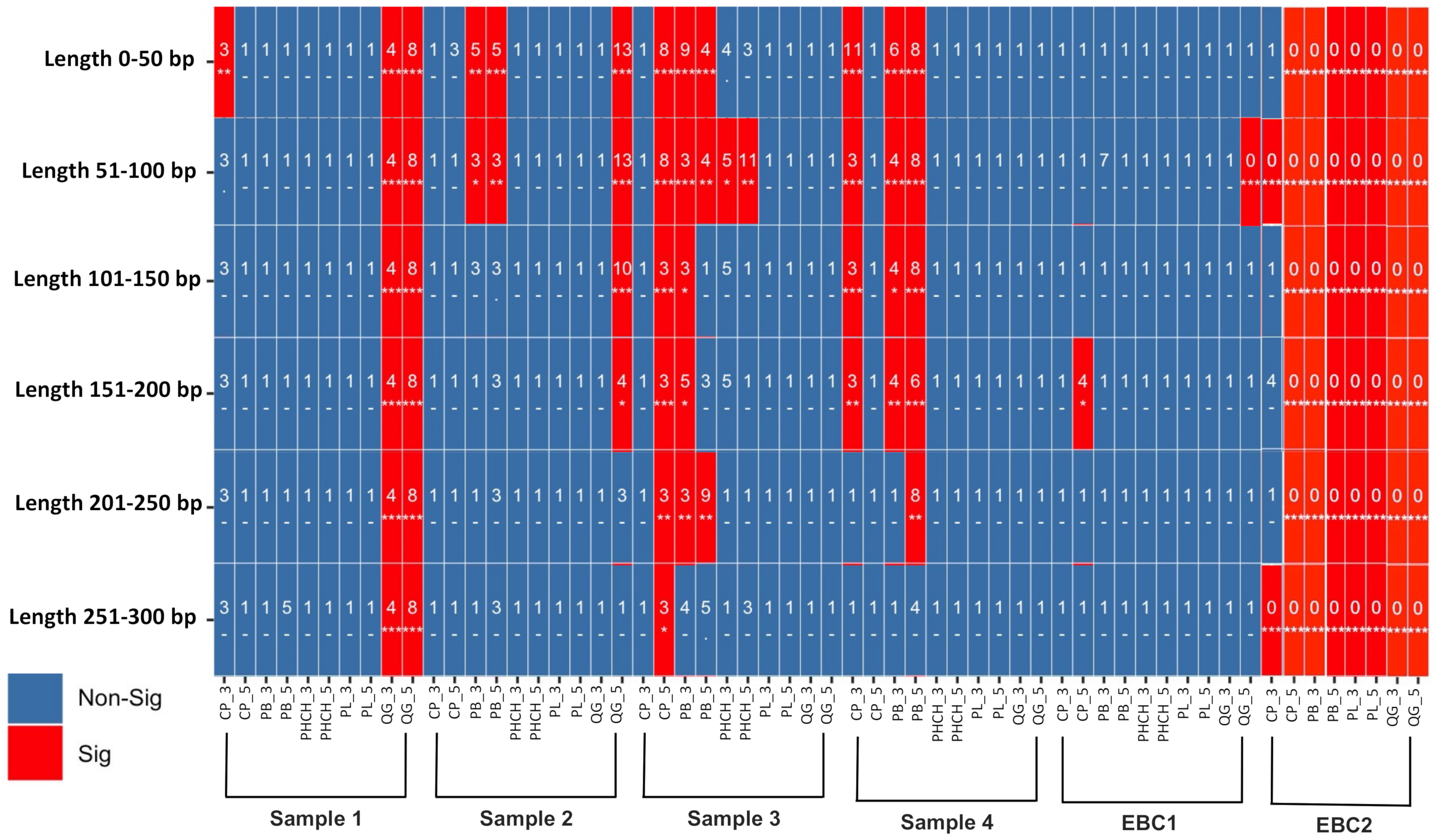
3.3.2. Effect of Contaminant Filtering
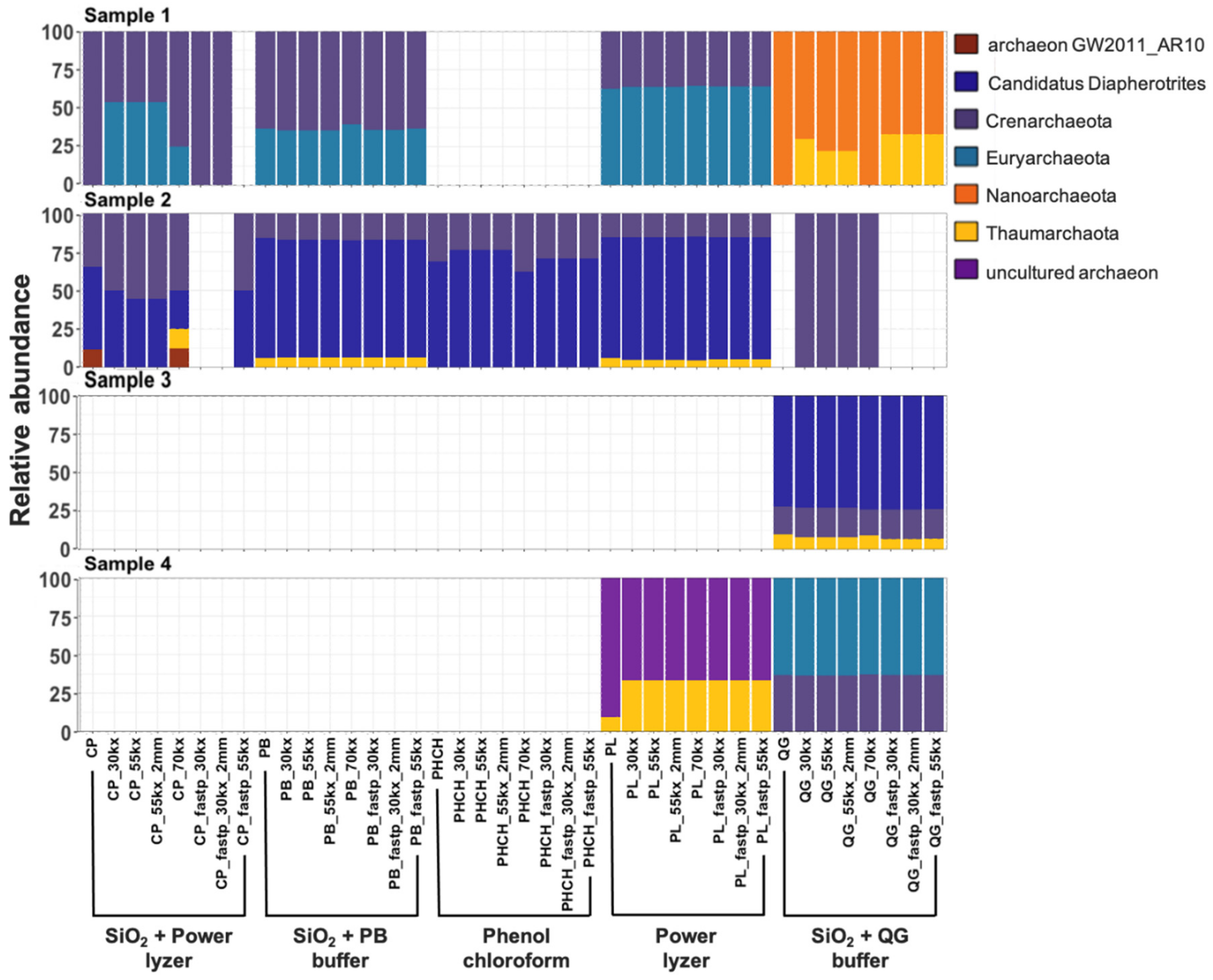
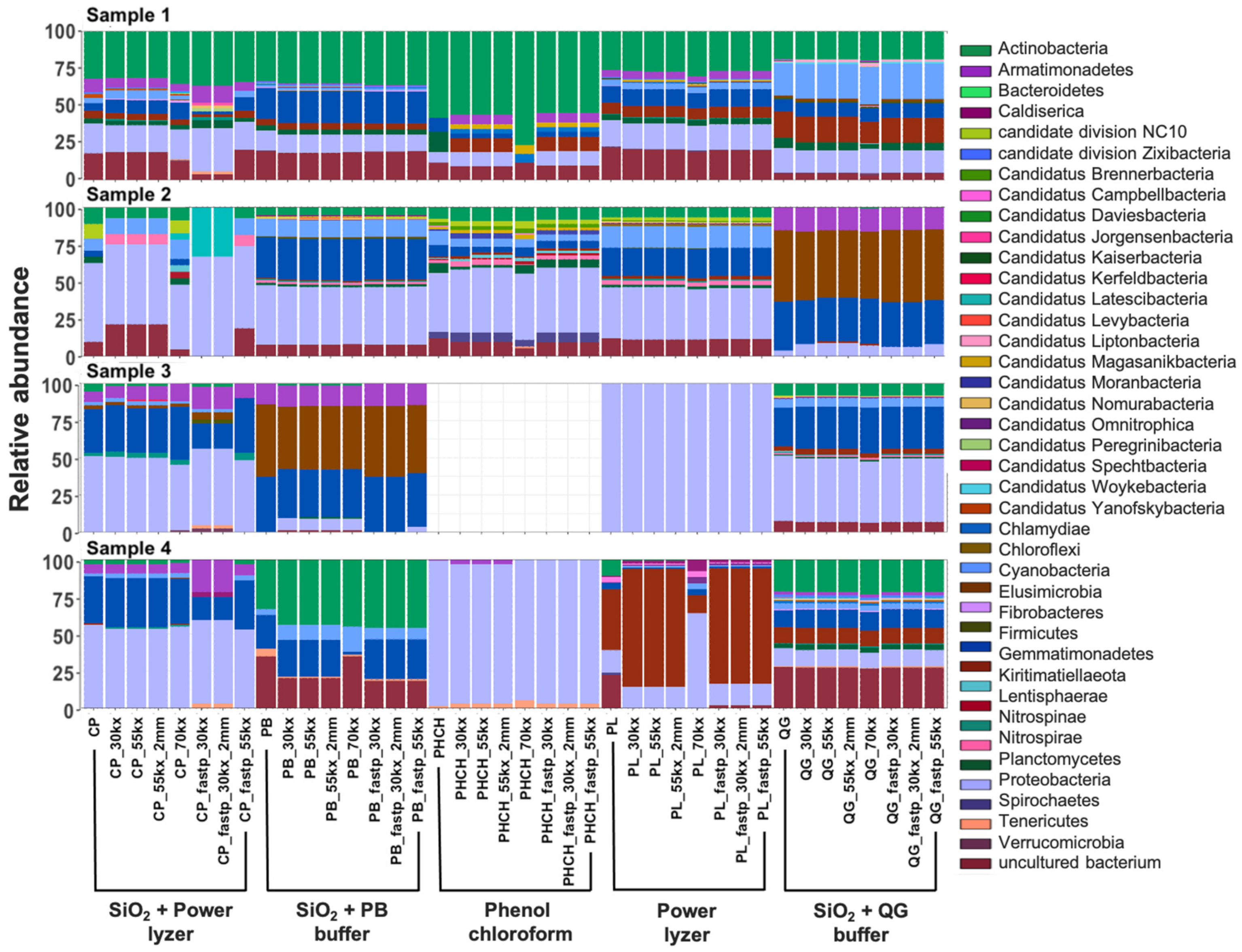
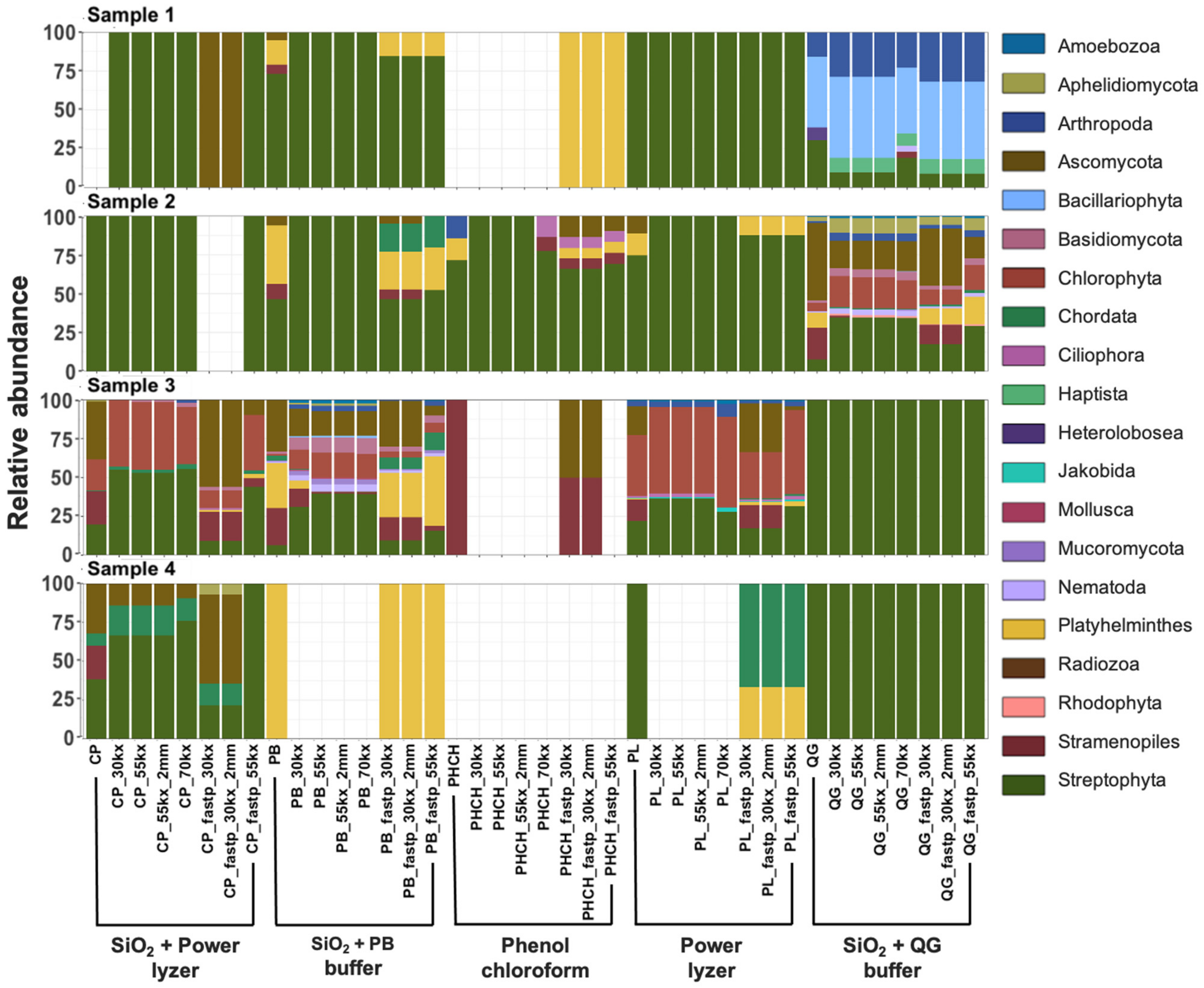
3.3.3. Taxonomic Biases of DNA Extractions Methods
3.3.4. Bioinformatic Preprocessing Has Minimal Impacts on Overall Prokaryotic Taxonomic Profiles
3.3.5. Impact of Reference Database Selection on Species Recovery
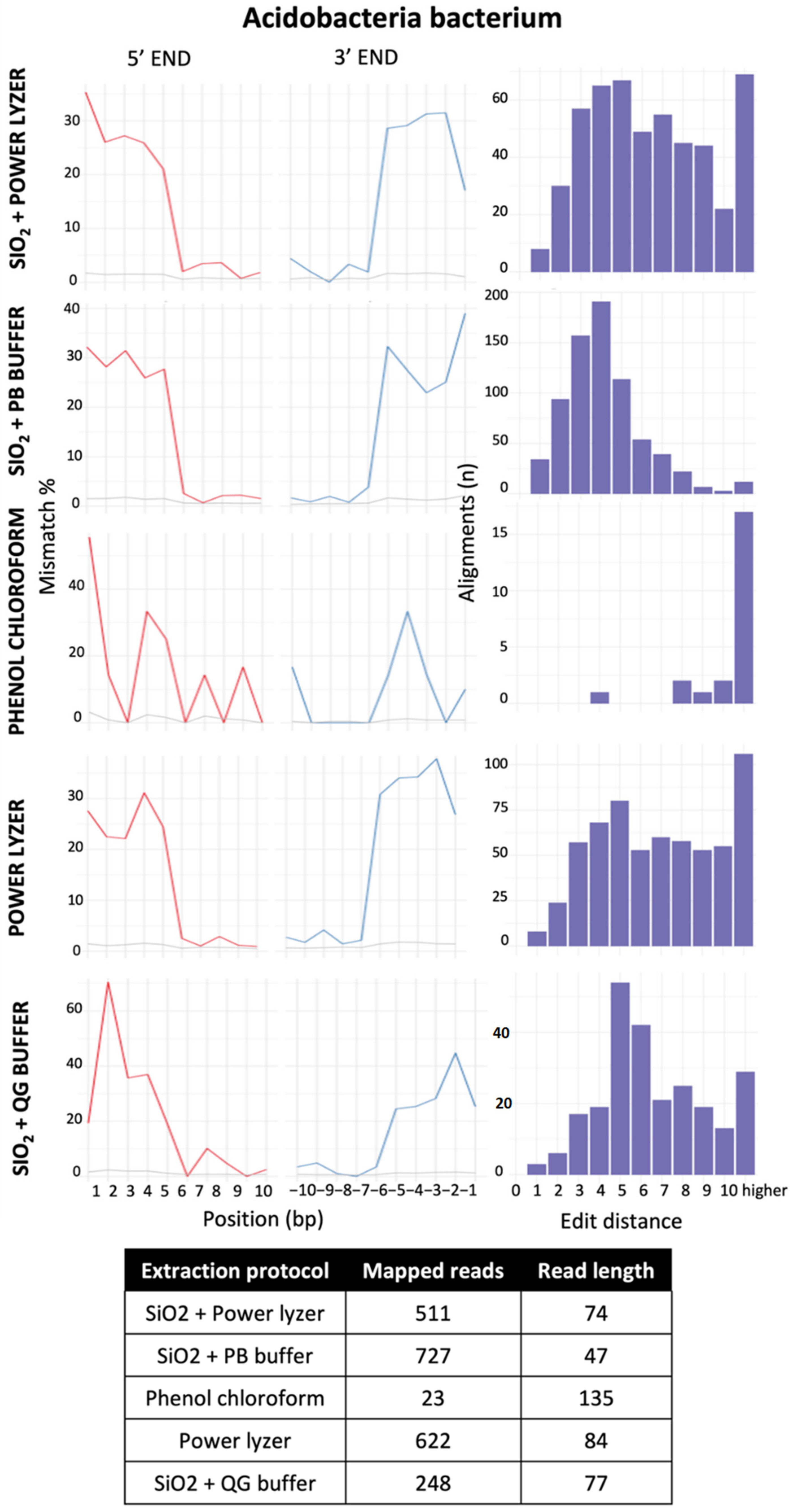
3.4. Environmental DNA Pool: Authenticating the aDNA Signal
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorgé, O.; Bennett, E.A.; Massilani, D.; Daligault, J.; Pruvost, M.; Geigl, E.M.; Grange, T. Analysis of Ancient DNA in Microbial Ecology. Methods Mol. Biol. 2016, 1399, 289–315. [Google Scholar] [PubMed]
- Liu, Y.; Weyrich, L.S.; Llamas, B. More Arrows in the Ancient DNA Quiver: Use of Paleoepigenomes and Paleomicrobiomes to Investigate Animal Adaptation to Environment. Mol. Biol. Evol. 2020, 37, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Arriola, L.A.; Cooper, A.; Weyrich, L.S. Palaeomicrobiology: Application of Ancient DNA Sequencing to Better Understand Bacterial Genome Evolution and Adaptation. Front. Ecol. Evol. 2020, 8, 40. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA analysis. Nat. Rev. Methods Primers 2021, 1, 14. [Google Scholar] [CrossRef]
- Yergeau, E.; Hogues, H.; Whyte, L.; Charles, G. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 2010, 4, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Slon, V.; Hopfe, C.; Weiß, C.L.; Mafessoni, F.; de la Rasilla, M.; Lalueza-Fox, C.; Rosas, A.; Soressi, M.; Knul, M.V.; Miller, R.; et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 2017, 356, 605–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frindte, K.; Lehndorff, E.; Vlaminck, S.; Werner, K.; Kehl, M.; Khormali, F.; Knief, C. Evidence for signatures of ancient microbial life in paleosols. Sci. Rep. 2020, 10, 16830. [Google Scholar] [CrossRef]
- Capo, E.; Giguet-Covex, C.; Rouillard, A.; Nota, K.; Heintzman, P.D.; Vuillemin, A.; Ariztegui, D.; Arnaud, F.; Belle, S.; Bertilsson, S.; et al. Lake Sedimentary DNA Research on Past Terrestrial and Aquatic Biodiversity: Overview and Recommendations. Quaternary 2021, 4, 6. [Google Scholar] [CrossRef]
- Frisia, S.; Weyrich, L.S.; Hellstrom, J.; Borsato, A.; Golledge, N.R.; Anesio, A.M. The influence of antarctic subglacial volcanism on the global iron cycle during the last glacial maximum. Nat. Commun. 2017, 8, 15425. [Google Scholar] [CrossRef] [Green Version]
- Turney, C.; Fogwill, C.J.; Golledge, N.R.; McKay, N.P.; van Sebille, E.; Jones, R.T.; Etheridge, D.; Rubino, M.; Thornton, D.P.; Davies, S.M.; et al. Early Last Interglacial ocean warming drove substantial ice mass loss from Antarctica. Proc. Natl. Acad. Sci. USA 2020, 117, 3996–4006. [Google Scholar] [CrossRef] [Green Version]
- Thomas, Z.A.; Mooney, S.; Cadd, H.; Baker, A.; Turney, C.; Schneider, L.; Hogg, A.; Haberle, S.; Green, K.; Weyrich, L.S.; et al. Late Holocene climate anomaly concurrent with fire activity and ecosystem shifts in the eastern Australian Highlands. Sci. Total Environ. 2020, 802, 149542. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Weyrich, L.S.; Clarke, L.J.; Cooper, A. Residual soil DNA extraction increases the discriminatory power between samples. Forensic Sci. Med. Pathol. 2015, 11, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Weyrich, L.S. Proper Authentication of Ancient DNA Is Still Essential. Genes 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armbrecht, L.J.; Coolen, M.J.L.; Lejzerowicz, F.; George, S.C.; Negandhi, K.; Suzuki, Y.; Young, J.; Foster, N.R.; Armand, L.K.; Cooper, A.; et al. Ancient DNA from marine sediments: Precautions and considerations for seafloor coring, sample handling and data generation. Earth Sci. Rev. 2019, 196, 102887. [Google Scholar] [CrossRef]
- Hagan, R.W.; Hofman, C.A.; Hübner, A.; Reinhard, K.; Schnorr, S.; Lewis, C.M., Jr.; Sankaranarayanan, K.; Warinner, C.G. Comparison of extraction methods for recovering ancient microbial DNA from paleofeces. Am. J. Phys. Anthropol. 2020, 171, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Warinner, C.; Herbig, A.; Mann, A.; Fellows Yates, J.A.; Weiß, C.L.; Burbano, H.A.; Orlando, L.; Krause, J. A Robust Framework for Microbial Archaeology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 321–356. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef]
- Velsko, I.M.; Frantz, L.; Herbig, A.; Larson, G.; Warinner, C. Selection of Appropriate Metagenome Taxonomic Classifiers for Ancient Microbiome Research. mSystems 2018, 3, e00080-18. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, R.; Weyrich, L.S. Assessing alignment-based taxonomic classification of ancient microbial DNA. PeerJ 2019, 7, e6594. [Google Scholar] [CrossRef] [Green Version]
- Ziesemer, K.A.; Mann, A.E.; Sankaranarayanan, K.; Schroeder, H.; Ozga, A.T.; Brandt, B.W.; Zaura, E.; Waters-Rist, A.; Hoogland, M.; Salazar-García, D.C.; et al. Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification. Sci. Rep. 2015, 5, 16498. [Google Scholar] [CrossRef]
- Capo, E.; Monchamp, M.E.; Coolen, M.J.L.; Domaizon, I.; Armbrecht, L.; Bertilsson, S. Environmental paleomicrobiology: Using DNA preserved in aquatic sediments to its full potential. Environ. Microbiol. 2022, 24, 2201–2209. [Google Scholar] [CrossRef]
- Nayfach, S.; Rodriguez-Mueller, B.; Garud, N.; Pollard, K.S. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 2016, 26, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Farmer, J.D. Hydrothermal systems: Doorways to early biosphere evolution. GSA Today 2020, 10, 1–9. [Google Scholar]
- Tassi, F.; Aguilera, F.; Darrah, T.; Vaselli, O.; Capaccioni, B.; Poreda, R.J.; Delgado Huertas, A. Fluid geochemistry of hydrothermal systems in the Arica-Parinacota, Tarapacá and Antofagasta regions (northern Chile). J. Volcanol. Geotherm. Res. 2010, 192, 1–15. [Google Scholar] [CrossRef]
- Procesi, M.; Cantucci, B.; Buttinelli, M.; Armezzani, G.; Quattrocchi, F.; Boschi, E. Strategic use of the underground in an Energy mix plan: Synergies among CO2, CH4 geological storage and geothermal energy. Latium Region case study (Central Italy). Appl. Energy 2014, 110, 104–131. [Google Scholar] [CrossRef]
- Scott, S.; Dorador, C.; Oyanedel, J.P.; Tobar, I.; Hengst, M.; Maya, G.; Harrod, C.; Vila, I. Microbial diversity and trophic components of two high altitude wetlands of the Chilean Altiplano. Gayana 2015, 79, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Risacher, F.; Alonso, H.; Salazar, C. The origin of brines and salts in Chilean salars: A hydrochemical review. Earth Sci. Rev. 2003, 63, 249–293. [Google Scholar] [CrossRef]
- Dorador, C.; Molina, V.; Hengst, M.; Eissler, Y.; Cornejo, M.; Fernández, C.; Pérez, V. Microbial Communities Composition, Activity, and Dynamics at Salar de Huasco: A Polyextreme Environment in the Chilean Altiplano. In Microbial Ecosystems in Central Andes Extreme Environments; Farías, M., Ed.; Springer: Cham, Switzerland, 2020; pp. 123–139. [Google Scholar]
- Hernández, K.L.; Yannicelli, B.; Olsen, L.M.; Dorador, C.; Menschel, E.J.; Molina, V.; Remonsellez, F.; Hengst, M.B.; Jeffrey, W.H. Microbial Activity Response to Solar Radiation across Contrasting Environmental Conditions in Salar de Huasco, Northern Chilean Altiplano. Front. Microbiol. 2016, 7, 1857. [Google Scholar] [CrossRef] [Green Version]
- Pérez, V.; Cortés, J.; Marchant, F.; Dorador, C.; Molina, V.; Cornejo-D’Ottone, M.; Hernández, K.; Jeffrey, W.; Barahona, S.; Hengst, M.B. Aquatic Thermal Reservoirs of Microbial Life in a Remote and Extreme High Andean Hydrothermal System. Microorganisms 2020, 8, 208. [Google Scholar] [CrossRef] [Green Version]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. Sci. Technol. Archaeol. Res. 2017, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fornari, M.; Risacher, F.; Féraud, G. Dating of paleolakes in the central Altiplano of Bolivia. Palaeogeogr. Palaeoclim. Palaeoecol. 2001, 172, 269–282. [Google Scholar] [CrossRef]
- Armbrecht, L.; Herrando-Pérez, S.; Eisenhofer, R.; Hallegraeff, G.M.; Bolch, C.; Cooper, A. An optimized method for the extraction of ancient eukaryote DNA from marine sediments. Mol. Ecol. 2020, 20, 906–919. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [Green Version]
- Gavrilov, S.N.; Stracke, C.; Jensen, K.; Menzel, P.; Kallnik, V.; Slesarev, A.; Sokolova, T.; Zayulina, K.; Bräsen, C.; Bonch-Osmolovskaya, E.A.; et al. Isolation and characterization of the first xylanolytic hyperthermophilic euryarchaeon Thermococcus sp. strain 2319x1 and its unusual multidomain glycosidase. Front. Microbiol. 2016, 7, 552. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hu, J.; Tian, H.; Li, S.; Liu, J.; Suzuki, M. A Low-Complexity GA-WSF Algorithm for Narrow-Band DOA Estimation. Int. J. Antennas Propag. 2018, 6, 7175653. [Google Scholar] [CrossRef]
- Clarke, E.L.; Taylor, L.J.; Zhao, C.; Connell, A.; Lee, J.J.; Fett, B.; Bushman, F.D.; Bittinger, K. Sunbeam: An extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 2019, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. A Quality Control Tool for High Throughput Sequence Data (2010). Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 July 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.E.; Sabin, S.; Ziesemer, K.; Vågene, Å.J.; Schroeder, H.; Ozga, A.T.; Sankaranarayanan, K.; Hofman, C.A.; Yates, J.A.F.; Salazar-García, D.C.; et al. Differential preservation of endogenous human and microbial DNA in dental calculus and dentin. Sci. Rep. 2018, 8, 9822. [Google Scholar] [CrossRef]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv 2016. preprint. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2013, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Hübler, R.; Key, F.M.; Warinner, C.; Bos, K.I.; Krause, J.; Herbig, A. HOPS: Automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol. 2019, 20, 280. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. The Role of Epigenetic Modifications and Microbiome Evolution in Bovid Adaptation to Environmental Changes. Ph.D. Thesis, The University of Adelaide, Adelaide, Australia, 2019. Available online: https://hdl.handle.net/2440/120689 (accessed on 4 October 2020).
- Yang, D.Y.; Watt, K. Contamination controls when preparing archaeological remains for ancient DNA analysis. J. Archaeol. Sci. 2005, 32, 331–336. [Google Scholar] [CrossRef]
- Pilli, E.; Modi, A.; Serpico, C.; Achilli, A.; Lancioni, H.; Lippi, B.; Bertoldi, F.; Gelichi, S.; Lari, M.; Caramelli, D. Monitoring DNA contamination in handled vs. directly excavated ancient human skeletal remains. PLoS ONE 2013, 8, e52524. [Google Scholar] [CrossRef]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do it right or not at all. Science 2000, 289, 1139. [Google Scholar] [CrossRef]
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Despres, V.; Hebler, J.; Rohland, N.; Kuch, M.; Krause, J.; Vigilant, L.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [Green Version]
- Champlot, S.; Berthelot, C.; Pruvost, M.; Bennett, E.A.; Grange, T.; Geigl, E.M. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE 2010, 5, e13042. [Google Scholar] [CrossRef] [Green Version]
- Knapp, M.; Clarke, A.C.; Horsburgh, K.A.; Matisoo-Smith, E.A. Setting the stage—Building and working in an ancient DNA laboratory. Ann. Anat. 2012, 194, 3–6. [Google Scholar] [CrossRef]
- Fulton, T.L.; Shapiro, B. Setting Up an Ancient DNA Laboratory. Methods Mol. Biol. 2019, 1963, 1–13. [Google Scholar]
- Weyrich, L.S.; Farrer, A.G.; Eisenhofer, R.; Arriola, L.A.; Young, J.; Selway, C.A.; Handsley-Davis, M.; Adler, C.J.; Breen, J.; Cooper, A. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 2019, 19, 982–996. [Google Scholar] [CrossRef] [Green Version]
- Ou, C.Y.; Moore, J.L.; Schochetman, G. Use of UV irradiation to reduce false positivity in polymerase chain reaction. BioTechniques 1991, 10, 442–446. [Google Scholar]
- Korlević, P.; Talamo, S.; Meyer, M. A combined method for DNA analysis and radiocarbon dating from a single sample. Sci. Rep. 2018, 8, 4127. [Google Scholar] [CrossRef] [Green Version]
- Farrer, A.G.; Wright, S.L.; Skelly, E.; Eisenhofer, R.; Dobney, K.; Weyrich, L.S. Effectiveness of decontamination protocols when analyzing ancient DNA preserved in dental calculus. Sci. Rep. 2021, 11, 7456. [Google Scholar] [CrossRef]
- Barbato, R.A.; Garcia-Reyero, N.; Foley, K.; Jones, R.; Courville, Z.; Douglas, T.; Perkins, E.; Reynolds, C.M. Removal of Exogenous Materials from the Outer Portion of Frozen Cores to Investigate the Ancient Biological Communities Harbored Inside. J. Vis. Exp. 2016, 113, 54091. [Google Scholar] [CrossRef]
- Saidi-Mehrabad, A.; Neuberger, P.; Cavaco, M.; Froese, D.; Lanoil, B. Optimization of subsampling, decontamination, and DNA extraction of difficult peat and silt permafrost samples. Sci. Rep. 2020, 10, 14295. [Google Scholar] [CrossRef]
- Orcutt, B.N.; Bergenthal, M.; Freudenthal, T.; Smith, D.; Lilley, M.D.; Schnieders, L.; Green, S.; Früh-Green, G.L. Contamination tracer testing with seabed drills: IODP Expedition 357. Sci. Dril. 2017, 23, 39–46. [Google Scholar] [CrossRef]
- Peigné, J.; Vian, J.; Cannavacciuolo, M.; Bottollier, B.; Chaussod, R. Soil sampling based on field spatial variability of soil microbial indicators. Eur. J. 2009, 45, 488–495. [Google Scholar] [CrossRef]
- Aguilar, P.; Acosta, E.; Dorador, C.; Sommaruga, R. Large Differences in Bacterial Community Composition among Three Nearby Extreme Waterbodies of the High Andean Plateau. Front. Microbiol. 2016, 7, 976. [Google Scholar] [CrossRef]
- Tecon, R.; Or, D. Biophysical processes supporting the diversity of microbial life in soil. FEMS Microbiol. Rev. 2017, 41, 599–623. [Google Scholar] [CrossRef]
- Penton, C.R.; Gupta, V.V.; Yu, J.; Tiedje, J.M. Size Matters: Assessing Optimum Soil Sample Size for Fungal and Bacterial Community Structure Analyses Using High Throughput Sequencing of rRNA Gene Amplicons. Front. Microbiol. 2016, 7, 824. [Google Scholar] [CrossRef]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Willerslev, E.; Cooper, A. Ancient DNA. Proc. Biol. 2005, 272, 3–16. [Google Scholar]
- McLaren, M.R.; Willis, A.D.; Callahan, B.J. Consistent and correctable bias in metagenomic sequencing experiments. eLife 2019, 8, e46923. [Google Scholar] [CrossRef]
- Aagaard, K.; Petrosino, J.; Keitel, W.; Watson, M.; Katancik, J.; Garcia, N.; Patel, S.; Cutting, M.; Madden, T.; Hamilton, H.; et al. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 2013, 27, 1012–1022. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotz, L.; Schwartz, T.; Thompson, L.; Humphrey, G.; Gogul, G.; Gaffney, J.; Humphrey, G.; Gogul, G.; Gaffney, J.; Amir, A.; et al. Earth Microbiome Project (EMP) High Throughput (HTP) DNA Extraction Protocol. Available online: https://www.protocols.io/view/earth-microbiome-project-emp-high-throughput-htp-d-8epv5qqjv1bz/v1 (accessed on 1 December 2021).
- Adler, C.J.; Dobney, K.; Weyrich, L.S.; Kaidonis, J.; Walker, A.W.; Haak, W. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the neolithic and industrial revolutions. Nat. Genet. 2013, 45, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warinner, C.; Rodrigues, J.F.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddymen, S.; et al. Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 2014, 46, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuillemin, A.; Ariztegui, D.; Nobbe, G.; Schubert, C.J.; PASADO Science Team. Influence of methanogenic populations in Holocene lacustrine sediments revealed by clone libraries and fatty acid biogeochemistry. GeoMicrobiol. J. 2013, 50, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Vuillemin, A.; Horn, F.; Alawi, M.; Henny, C.; Wagner, D.; Crowe, S.; Kallmeyer, J. Preservation and significance of extracellular DNA in ferruginous sediments from Lake Towuti, Indonesia. Front. Microbiol. 2017, 8, 1440. [Google Scholar] [CrossRef] [Green Version]
- Vuillemin, A.; Horn, F.; Friese, A.; Winkel, M.; Alawi, M.; Wagner, D.; Henny, C.; Orsi, W.D.; Crowe, S.A.; Kallmeyer, J. Metabolic potential of microbial communities from ferruginous sediments. Environ. Microbiol. 2018, 20, 4297–4313. [Google Scholar] [CrossRef]
- Kircher, M. Analysis of high-throughput ancient DNA sequencing data. Methods Mol. Biol. 2012, 840, 197–228. [Google Scholar]
- Schubert, M.; Ermini, L.; Der Sarkissian, C.; Jónsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.D.; Fernández, R.; Kircher, M.; McCue, M.; et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef]
- Kircher, M.; Heyn, P.; Kelso, J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genom. 2011, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Rokas, A. Prevention, diagnosis and treatment of high-throughput sequencing data pathologies. Mol. Ecol. 2014, 23, 1679–1700. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Arizmendi Cárdenas, Y.O.; Neuenschwander, S.; Malaspinas, A.S. Benchmarking metagenomics classifiers on ancient viral DNA: A simulation study. PeerJ 2022, 10, e12784. [Google Scholar] [CrossRef]
- Mann, A.E.; Fellows Yates, J.A.; Fagernäs, Z.; Austin, R.M.; Nelson, E.A.; Hofman, C.A. Do I have something in my teeth? The trouble with genetic analyses of diet from archaeological dental calculus. Quat. Int. 2020, in press. [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Yu, W.H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, 2010, baq013. [Google Scholar] [CrossRef]
- Obregon-Tito, A.J.; Tito, R.Y.; Metcalf, J.; Sankaranarayanan, K.; Clemente, J.C.; Ursell, L.K.; Xu, Z.Z.; Van Treuren, W.; Knight, R.; Gaffney, P.M.; et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015, 6, 6505. [Google Scholar] [CrossRef] [Green Version]
- Wibowo, M.C.; Yang, Z.; Borry, M.; Hübner, A.; Huang, K.D.; Tierney, B.T.; Zimmerman, S.; Barajas-Olmos, F.; Contreras-Cubas, C.; García-Ortiz, H.; et al. Reconstruction of ancient microbial genomes from the human gut. Nature 2021, 594, 234–239. [Google Scholar] [CrossRef]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth, and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef]
- Acosta, O.; Custodio, E. Impactos ambientales de las extracciones de agua subterránea en el Salar del Huasco (norte de Chile). Bol. Geol. Min. 2008, 119, 33–50. [Google Scholar]
- Alvarez, R.; Lavado, R.S. Climate, Organic Matter and Clay Content Relationships in the Pampa and Chaco Soils, Argentina. Geoderma 1998, 83, 127–141. [Google Scholar] [CrossRef]
- Sand, K.K.; Jelavić, S. Mineral Facilitated Horizontal Gene Transfer: A New Principle for Evolution of Life? Front. Microbiol. 2018, 9, 2217. [Google Scholar] [CrossRef] [Green Version]
- Zainabadi, K.; Nyunt, M.M.; Plowe, C.V. An improved nucleic acid extraction method from dried blood spots for amplification of Plasmodium falciparum kelch13 for detection of artemisinin resistance. Malar. J. 2019, 18, 192. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.W.; Overballe-Petersen, S.; Ermini, L.; Sarkissian, C.D.; Haile, J.; Hellstrom, M.; Spens, J.; Thomsen, P.F.; Bohmann, K.; Cappellini, E.; et al. Ancient and modern environmental DNA. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinegger, M.; Salzberg, S.L. Terminating contamination: Large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 2020, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Delihas, N. Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 2011, 3, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Pääbo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar] [CrossRef] [Green Version]
- Schuenemann, V.J.; Bos, K.; DeWitte, S.; Schmedes, S.; Jamieson, J.; Mittnik, A.; Forrest, S.; Coombes, B.K.; Wood, J.W.; Earn, D.J.D.; et al. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proc. Natl. Acad. Sci. USA 2011, 108, E746–E752. [Google Scholar] [CrossRef] [Green Version]
- Borry, M.; Hübner, A.; Rohrlach, A.B.; Warinner, C. PyDamage: Automated ancient damage identification and estimation for contigs in ancient DNA de novo assembly. PeerJ 2021, 9, e11845. [Google Scholar] [CrossRef]
- Neukamm, J.; Peltzer, A.; Nieselt, K. DamageProfiler: Fast damage pattern calculation for ancient DNA. Bioinformatics 2021, 37, 3652–3653. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, V.; Liu, Y.; Hengst, M.B.; Weyrich, L.S. A Case Study for the Recovery of Authentic Microbial Ancient DNA from Soil Samples. Microorganisms 2022, 10, 1623. https://doi.org/10.3390/microorganisms10081623
Pérez V, Liu Y, Hengst MB, Weyrich LS. A Case Study for the Recovery of Authentic Microbial Ancient DNA from Soil Samples. Microorganisms. 2022; 10(8):1623. https://doi.org/10.3390/microorganisms10081623
Chicago/Turabian StylePérez, Vilma, Yichen Liu, Martha B. Hengst, and Laura S. Weyrich. 2022. "A Case Study for the Recovery of Authentic Microbial Ancient DNA from Soil Samples" Microorganisms 10, no. 8: 1623. https://doi.org/10.3390/microorganisms10081623