
In Situ Genomics and Transcriptomics of SAR202 Subclusters Revealed Subtle Distinct Activities in Deep-Sea Water


Abstract
:1. Introduction
2. Material and Methods
2.1. In Situ Nucleic Acid Extraction
2.2. Omics Sequencing
2.3. Data Analysis
2.4. Annotation and Phylogenomics of SAR202 MAGs
2.5. Relative Abundance of the SAR202 Subclusters in Metagenomics and Metatranscriptomics
2.6. Transcriptional Activities and Gene Expression Patterns of SAR202 Subclusters
3. Results
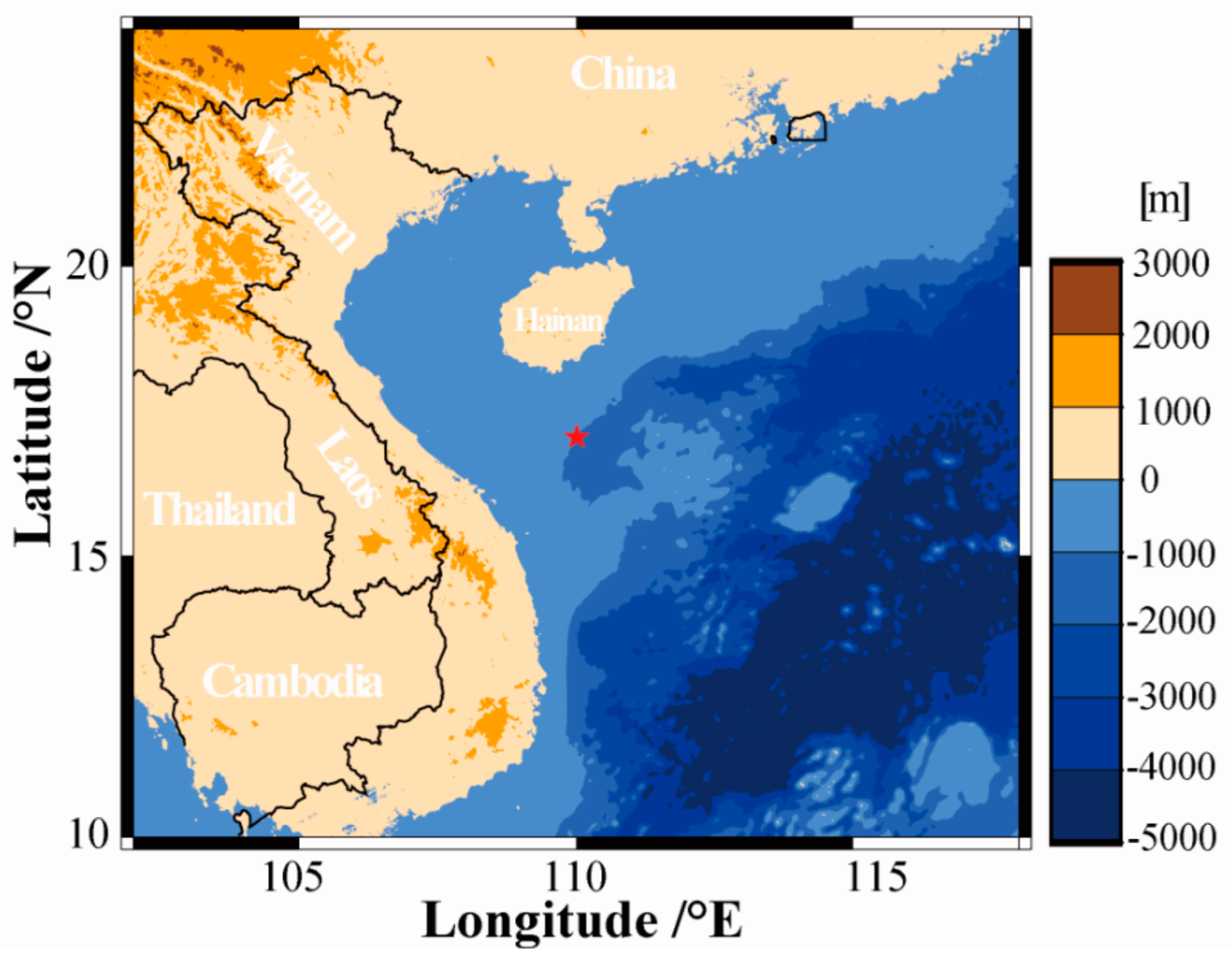
3.1. In Situ Samples’ Collection and Environmental Factors’ Measurement
3.2. Microbial Community in the Metagenomic and Metatranscriptomic Data
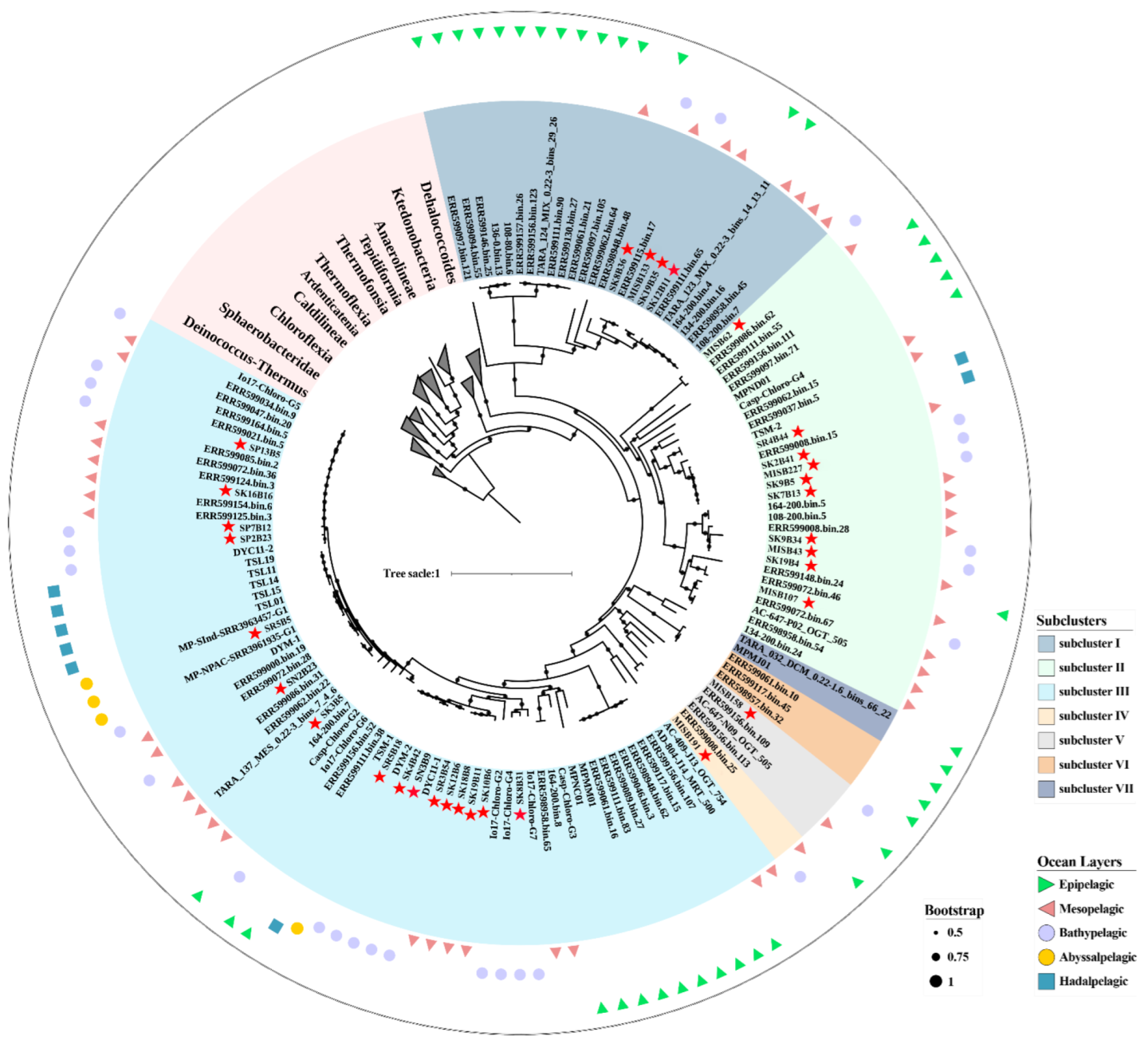
3.3. SAR202 Genome Reconstruction and Affinity Relationships
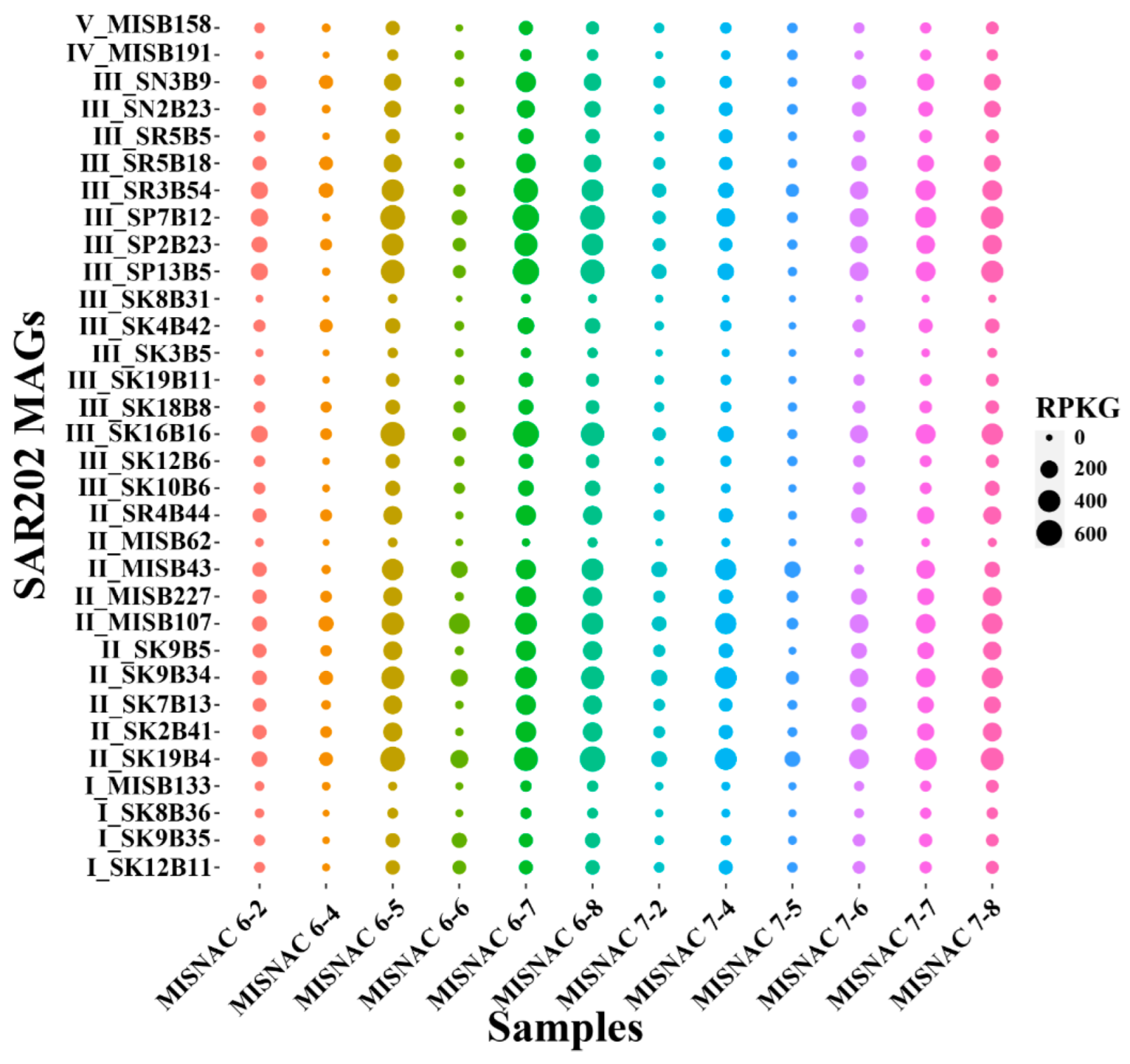
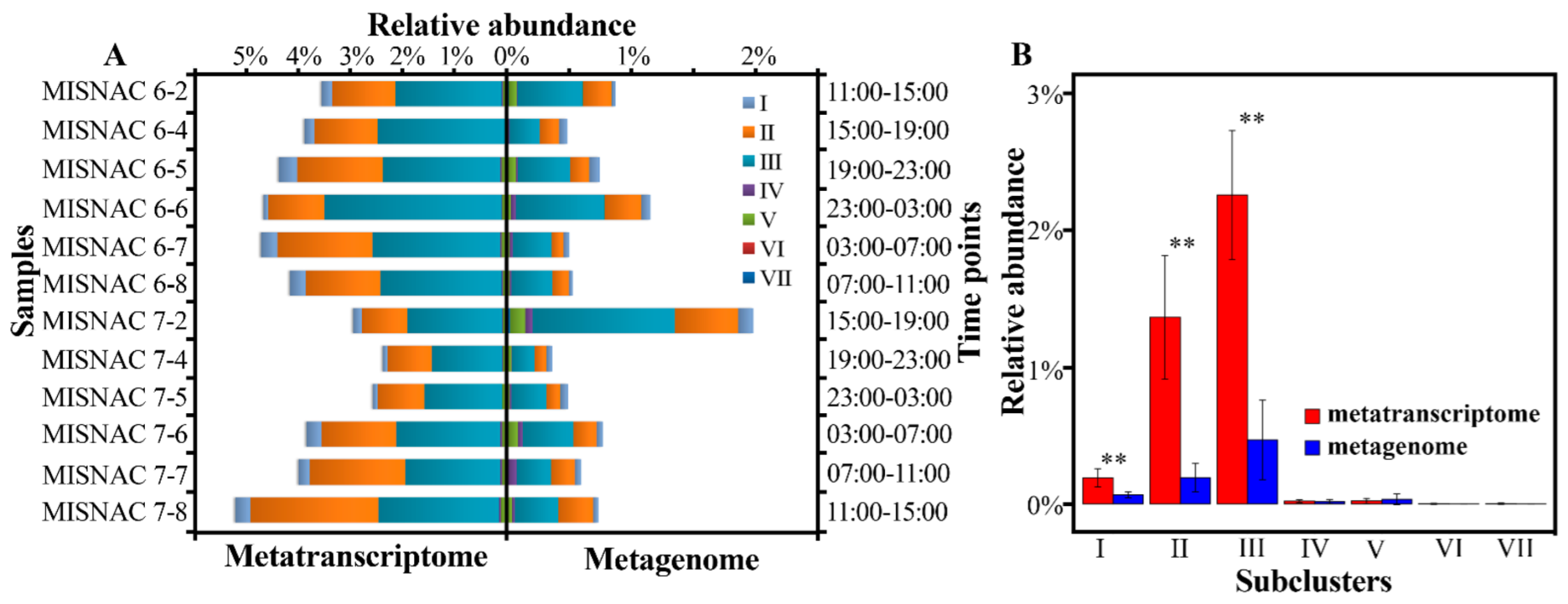
3.4. Transcriptional Activities of SAR202 Subclusters
3.5. Functional Gene Transcripts in SAR202 Subclusters
4. Discussion
4.1. Phylogenetic Analysis of SAR202
4.2. Time-Course Changes in SAR202 Subclusters
4.3. Carbon Sources for Each SAR202 Subcluster
4.4. Contribution of SAR202 Subclusters in Sulfur Metabolism
4.5. Roles of Each SAR202 Subcluster in Nitrogen Metabolism
4.6. Choice of Osmolytes by Each SAR202 Subcluster
4.7. Metabolism of Other Substances
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyer-Reil, L.A.; Thingstad, F. The Ecological Role of Water-Column Microbes in the Sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Arístegui, J.; Gasol, J.M.; Duarte, C.M.; Herndld, G.J. Microbial oceanography of the dark ocean’s pelagic realm. Limnol. Oceanogr. 2009, 54, 1501–1529. [Google Scholar] [CrossRef]
- Glover, A.G.; Gooday, A.J.; Bailey, D.M.; Billett, D.S.; Chevaldonné, P.; Colaço, A.; Copley, J.; Cuvelier, D.; Desbruyères, D.; Kalogeropoulou, V. Temporal change in deep-sea benthic ecosystems: A review of the evidence from recent time-series studies. Adv. Mar. Biol. 2010, 58, 1–95. [Google Scholar] [PubMed]
- Eloe, E.A.; Shulse, C.; Fadrosh, D.W.; Williamson, S.J.; Allen, E.E.; Bartlett, D.H. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 2010, 3, 449–458. [Google Scholar] [CrossRef]
- Gao, Z.; Huang, J.; Cui, G.; Li, W.; Li, J.; Wei, Z.; Chen, J.; Xin, Y.; Cai, D.; Zhang, A.; et al. In situ meta-omic insights into the community compositions and ecological roles of hadal microbes in the Mariana Trench. Environ. Microbiol. 2019, 21, 4092–4108. [Google Scholar] [CrossRef] [PubMed]
- Urbach, E.; Vergin, K.L.; Larson, G.L.; Giovannoni, S.J. Bacterioplankton communities of Crater Lake, OR: Dynamic changes with euphotic zone food web structure and stable deep water populations. Hydrobiologia 2007, 191, 161–177. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Cui, G.; Nunoura, T.; Takaki, Y.; Li, W.; Li, J.; Gao, Z.; Takai, K.; Zhang, A.; et al. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ. Microbiol. 2018, 21, 716–729. [Google Scholar] [CrossRef]
- Estrada, M.; Bayer-Giraldi, M.; Felipe, J.; Marrasé, C.; Sala, M.; Vidal, M. Light and nutrient effects on microbial communities collected during spring and summer in the Beaufort Sea. Aquat. Microb. Ecol. 2009, 54, 217–231. [Google Scholar] [CrossRef]
- Feike, J.; Jürgens, K.; Hollibaugh, J.T.; Krüger, S.; Jost, G.; Labrenz, M. Measuring unbiased metatranscriptomics in suboxic waters of the central Baltic Sea using a new in situ fixation system. ISME J. 2011, 6, 461–470. [Google Scholar] [CrossRef]
- Marietou, A.; Bartlett, D.H. Effects of High Hydrostatic Pressure on Coastal Bacterial Community Abundance and Diversity. Appl. Environ. Microbiol. 2014, 80, 5992–6003. [Google Scholar] [CrossRef]
- Wei, Z.-F.; Li, W.-L.; Li, J.; Chen, J.; Xin, Y.-Z.; He, L.-S.; Wang, Y. Multiple in situ Nucleic Acid Collections (MISNAC) from Deep-Sea Waters. Front. Mar. Sci. 2020, 7, 81. [Google Scholar] [CrossRef]
- Edgcomb, V.; Taylor, C.; Pachiadaki, M.; Honjo, S.; Engstrom, I.; Yakimov, M. Comparison of Niskin vs. in situ approaches for analysis of gene expression in deep Mediterranean Sea water samples. Deep Sea Res. Part II Top. Stud. Oceanogr. 2014, 129, 213–222. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Z.-M.; Li, J.; He, L.-S.; Cui, G.-J.; Li, W.-L.; Chen, J.; Xin, Y.-Z.; Cai, D.-S.; Zhang, A.-Q. Hadal water sampling by in situ microbial filtration and fixation (ISMIFF) apparatus. Deep Sea Res. Part I Oceanogr. Res. Pap. 2019, 144, 132–137. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Rappé, M.S.; Vergin, K.L.; Adair, N.L. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the Green Non-Sulfur bacteria. Proc. Natl. Acad. Sci. USA 1996, 93, 7979–7984. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-F.; Li, W.-L.; Huang, J.-M.; Wang, Y. Metagenomic studies of SAR202 bacteria at the full-ocean depth in the Mariana Trench. Deep Sea Res. Part I Oceanogr. Res. Pap. 2020, 165, 103396. [Google Scholar] [CrossRef]
- Morris, R.M.; Rappé, M.S.; Urbach, E.; Connon, S.A.; Giovannoni, S.J. Prevalence of the Chloroflexi-Related SAR202 Bacterioplankton Cluster throughout the Mesopelagic Zone and Deep Ocean. Appl. Environ. Microbiol. 2004, 70, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Landry, Z.; Swan, B.K.; Herndl, G.J.; Stepanauskas, R.; Giovannoni, S.J. SAR202 Genomes from the Dark Ocean Predict Pathways for the Oxidation of Recalcitrant Dissolved Organic Matter. mBio 2017, 8, e00413–e00417. [Google Scholar] [CrossRef]
- Saw, J.H.W.; Nunoura, T.; Hirai, M.; Takaki, Y.; Parsons, R.; Michelsen, M.; Longnecker, K.; Kujawinski, E.B.; Stepanauskas, R.; Landry, Z.; et al. Pangenomics Analysis Reveals Diversification of Enzyme Families and Niche Specialization in Globally Abundant SAR202 Bacteria. mBio 2020, 11, e02975-19. [Google Scholar] [CrossRef] [PubMed]
- Mehrshad, M.; Rodriguez-Valera, F.; Amoozegar, M.A.; López-García, P.; Ghai, R. The enigmatic SAR202 cluster up close: Shedding light on a globally distributed dark ocean lineage involved in sulfur cycling. ISME J. 2017, 12, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Qiong, W.; Orla, O.S.; Rachel, G.D.; Cole, J.R.; Paul, R.R.; O’Toole, P.W. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010, 38, e200. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC a Quality Control Tool for High Throughput Sequence Data. 2013. Available online: https://github.com/s-andrews/FastQC (accessed on 8 July 2020).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. FastUniq: A Fast De Novo Duplicates Removal Tool for Paired Short Reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gilna, P.; Li, W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009, 25, 1338–1340. [Google Scholar] [CrossRef]
- Ramiro, L.; Shinichi, S.; Guillem, S.; Cornejo-Castillo, F.M.; Isabel, F.; Hugo, S.; Pascal, H.; Hiroyuki, O.; Colomban, D.V.; Gipsi, L.M. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Hyatt, D.; Locascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; Mcminn, A.; Wang, M.; Xie, B.-B.; Qin, Q.-L.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Herndl, G.J.; Hansell, D.A.; Benner, R.; Kattner, G.; Wilhelm, S.W.; Kirchman, D.L.; Weinbauer, M.G.; Luo, T.; Chen, F.; et al. Microbial production of recalcitrant dissolved organic matter: Long-term carbon storage in the global ocean. Nat. Rev. Genet. 2010, 8, 593–599. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Bolin, J.T.; Eltis, L.D. Ring-Cleavage Dioxygenases; Springer: New York, NY, USA, 2004; pp. 359–395. [Google Scholar]
- Chen, Y.-X.; Liu, H.; Zhu, L.-C.; Jin, Y.-F. Cloning and characterization of a chromosome-encoded catechol 2,3-dioxygenase gene from Pseudomonas aeruginosa ZD 4-3. Microbiology 2004, 73, 689–695. [Google Scholar] [CrossRef]
- Cipollone, R.; Ascenzi, P.; Visca, P. Common themes and variations in the rhodanese superfamily. IUBMB Life 2007, 59, 51–59. [Google Scholar] [CrossRef]
- Aussignargues, C.; Giuliani, M.-C.; Infossi, P.; Lojou, E.; Guiral, M.; Giudici-Orticoni, M.-T.; Ilbert, M. Rhodanese Functions as Sulfur Supplier for Key Enzymes in Sulfur Energy Metabolism. J. Biol. Chem. 2012, 287, 19936–19948. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, S.; Kroder, M.; Dilling, W.; Cypionka, H. Oxidation of H2, organic compounds and inorganic sulfur compounds coupled to reduction of O2 or nitrate by sulfate-reducing bacteria. Arch. Microbiol. 1992, 158, 93–99. [Google Scholar] [CrossRef]
- Park, S.; Smith, L.T.; Smith, G.M. Role of Glycine Betaine and Related Osmolytes in Osmotic Stress Adaptation in Yersinia enterocolitica ATCC 9610. Appl. Environ. Microbiol. 1995, 61, 4378–4381. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H.; Rhea, M.D.; Kemp, K.M.; Bailey, D. Trimethylamine oxide, betaine and other osmolytes in deep-sea animals: Depth trends and effects on enzymes under hydrostatic pressure. Cell. Mol. Biol. 2004, 50, 371–376. [Google Scholar] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome ID | Depth (m) | Subcluster | Longitude (°E) | Latitude (°N) | Genome Size (bp) | GC (%) | CDS Number | Coding Density | Compl (%) | Conta (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| MISB158 | 1022 | V | 110.01 | 17.06 | 1,133,699 | 0.46 | 1299 | 0.88 | 50.69 | 6.89 |
| MISB191 | 1022 | IV | 110.01 | 17.06 | 2,050,173 | 0.56 | 2106 | 0.87 | 95.38 | 2.97 |
| SN3B9 | 1022 | III | 114.99 | 11.97 | 2,219,140 | 0.58 | 2398 | 0.85 | 53.21 | 3.47 |
| SN2B23 | 2000 | III | 116.49 | 11.99 | 2,277,881 | 0.59 | 2254 | 0.87 | 78.88 | 1.98 |
| SR5B5 | 4210 | III | 123.03 | 22.19 | 3,297,532 | 0.59 | 3180 | 0.86 | 95.71 | 0.99 |
| SR5B18 | 4210 | III | 123.03 | 22.19 | 3,974,242 | 0.58 | 3976 | 0.85 | 93.62 | 3.96 |
| SR3B54 | 1646 | III | 120.04 | 21.87 | 2,388,874 | 0.58 | 2416 | 0.85 | 77.43 | 2.15 |
| SP7B12 | 1000 | III | 110.17 | 17.19 | 1,461,202 | 0.57 | 1616 | 0.85 | 80.77 | 0.99 |
| SP2B23 | 1000 | III | 110.18 | 17.19 | 2,554,614 | 0.58 | 3004 | 0.87 | 57.49 | 4.13 |
| SP13B5 | 1000 | III | 110.17 | 17.19 | 1,973,705 | 0.57 | 2212 | 0.87 | 55.05 | 7.13 |
| SK8B31 | 3000 | III | 118.17 | 13.96 | 2,810,028 | 0.56 | 2809 | 0.84 | 67.21 | 0.61 |
| SK4B42 | 3000 | III | 116.48 | 18.01 | 2,736,876 | 0.58 | 2931 | 0.86 | 52.55 | 1.75 |
| SK3B5 | 200 | III | 116.48 | 18.01 | 2,415,447 | 0.55 | 2531 | 0.87 | 82.84 | 4.76 |
| SK19B11 | 500 | III | 118.2 | 16 | 3,020,336 | 0.57 | 3218 | 0.85 | 89.19 | 3.3 |
| SK18B8 | 500 | III | 118.18 | 14.98 | 2,560,045 | 0.57 | 2812 | 0.86 | 91.75 | 2.48 |
| SK16B16 | 500 | III | 114.99 | 9.99 | 2,946,073 | 0.57 | 2930 | 0.86 | 79.07 | 2.67 |
| SK12B6 | 500 | III | 114.99 | 11.97 | 2,493,422 | 0.57 | 2749 | 0.87 | 81.66 | 1.52 |
| SK10B6 | 600 | III | 116.49 | 14 | 1,833,071 | 0.57 | 1968 | 0.86 | 56.46 | 9.03 |
| SR4B44 | 6000 | II | 123.31 | 22.82 | 2,606,463 | 0.56 | 3020 | 0.86 | 98.68 | 0 |
| MISB62 | 1022 | II | 110.01 | 17.06 | 1,047,704 | 0.3 | 1167 | 0.94 | 93.73 | 2.97 |
| MISB43 | 1022 | II | 110.01 | 17.06 | 1,446,211 | 0.42 | 1609 | 0.92 | 67.87 | 1.65 |
| MISB227 | 1022 | II | 110.01 | 17.06 | 3,068,488 | 0.56 | 3200 | 0.86 | 61.73 | 1.1 |
| MISB107 | 1022 | II | 110.01 | 17.06 | 1,845,114 | 0.46 | 1919 | 0.9 | 78.11 | 6.53 |
| SK9B5 | 1000 | II | 116.49 | 14 | 2,676,491 | 0.56 | 2880 | 0.86 | 96.7 | 1.1 |
| SK9B34 | 1000 | II | 116.49 | 14 | 1,950,012 | 0.43 | 2185 | 0.9 | 92.24 | 1.98 |
| SK7B13 | 850 | II | 118.19 | 16.66 | 2,301,923 | 0.56 | 2574 | 0.86 | 61.95 | 0 |
| SK2B41 | 2000 | II | 116.48 | 18.01 | 3,383,583 | 0.56 | 3501 | 0.85 | 82.84 | 4.76 |
| SK19B4 | 500 | II | 118.2 | 16 | 2,083,146 | 0.43 | 2022 | 0.92 | 68.96 | 5.94 |
| MISB133 | 1022 | I | 110.01 | 17.06 | 2,386,853 | 0.58 | 2217 | 0.85 | 92.65 | 2.4 |
| SK8B36 | 3000 | I | 118.17 | 13.96 | 2,329,958 | 0.58 | 2238 | 0.85 | 69.08 | 0 |
| SK19B35 | 500 | I | 118.2 | 16 | 1,219,668 | 0.47 | 1243 | 0.9 | 68.28 | 1.98 |
| SK12B11 | 500 | I | 114.99 | 11.97 | 1,660,536 | 0.47 | 1736 | 0.9 | 54.32 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Z.; Li, Q.; Lu, R.; Zheng, P.; Wang, Y. In Situ Genomics and Transcriptomics of SAR202 Subclusters Revealed Subtle Distinct Activities in Deep-Sea Water. Microorganisms 2022, 10, 1629. https://doi.org/10.3390/microorganisms10081629
Wei Z, Li Q, Lu R, Zheng P, Wang Y. In Situ Genomics and Transcriptomics of SAR202 Subclusters Revealed Subtle Distinct Activities in Deep-Sea Water. Microorganisms. 2022; 10(8):1629. https://doi.org/10.3390/microorganisms10081629
Chicago/Turabian StyleWei, Zhanfei, Qingmei Li, Rui Lu, Pengfei Zheng, and Yong Wang. 2022. "In Situ Genomics and Transcriptomics of SAR202 Subclusters Revealed Subtle Distinct Activities in Deep-Sea Water" Microorganisms 10, no. 8: 1629. https://doi.org/10.3390/microorganisms10081629