

Simultaneous Metabarcoding and Quantification of Neocallimastigomycetes from Environmental Samples: Insights into Community Composition and Novel Lineages

 , , , , , , , ,
, , , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primer Design and Validation
2.2. Cultivation of Pure Cultures
2.3. Environmental Samples
2.4. DNA Extraction
2.5. Polymerase Chain Reaction (PCR)
2.6. Quantitative Real-Time PCR (qPCR)
2.7. Amplicon Sequencing
2.8. Mock Communities to Test the Data Processing Pipeline
2.9. Sequencing Data Processing
2.10. Phylogenetic Analysis
3. Results and Discussion
3.1. In Silico Primer Search
- (i)
- The gap in coverage of AGF is closed compared to earlier reported primer sets, e.g., AGF-LSU reverse [14] did not detect Aestipascuomyces, Agriosomyces, Buwchfawromyces, and Joblinomyces; GGNL1 forward [18] did not match with newly described AL3 and AL8 clades [15], and GGNL4 reverse did not match with Anaeromyces contortus.
- (ii)
- In contrast to earlier methods where detection, quantification, and classification needed several steps with different primers, here only one genetic marker is used, avoiding bias by different primer selectivities.
- (iii)
- The alienability of the D2 LSU amplicon presents advantages in comparison with any ITS amplicon and its intra-strain heterogeneity is much lower in the LSU than in the ITS regions where the differences frequently exceed species limits.
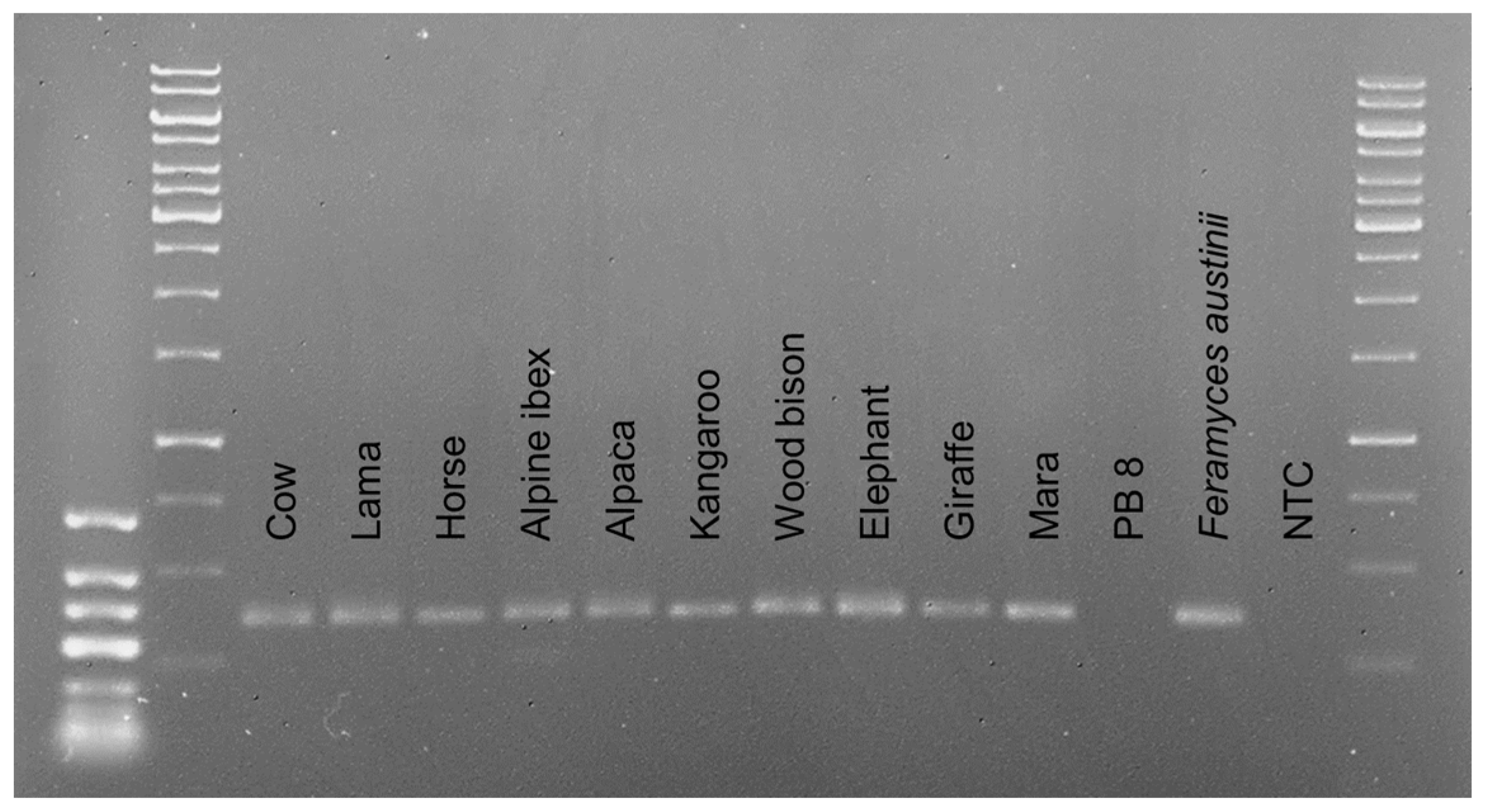
3.2. Detection of AGF in Cultures and Environmental Samples
3.3. Quantitative Real-Time PCR
3.4. Evaluation of the AGF-LSU-EnvS Processing Pipeline Using Mock Communities
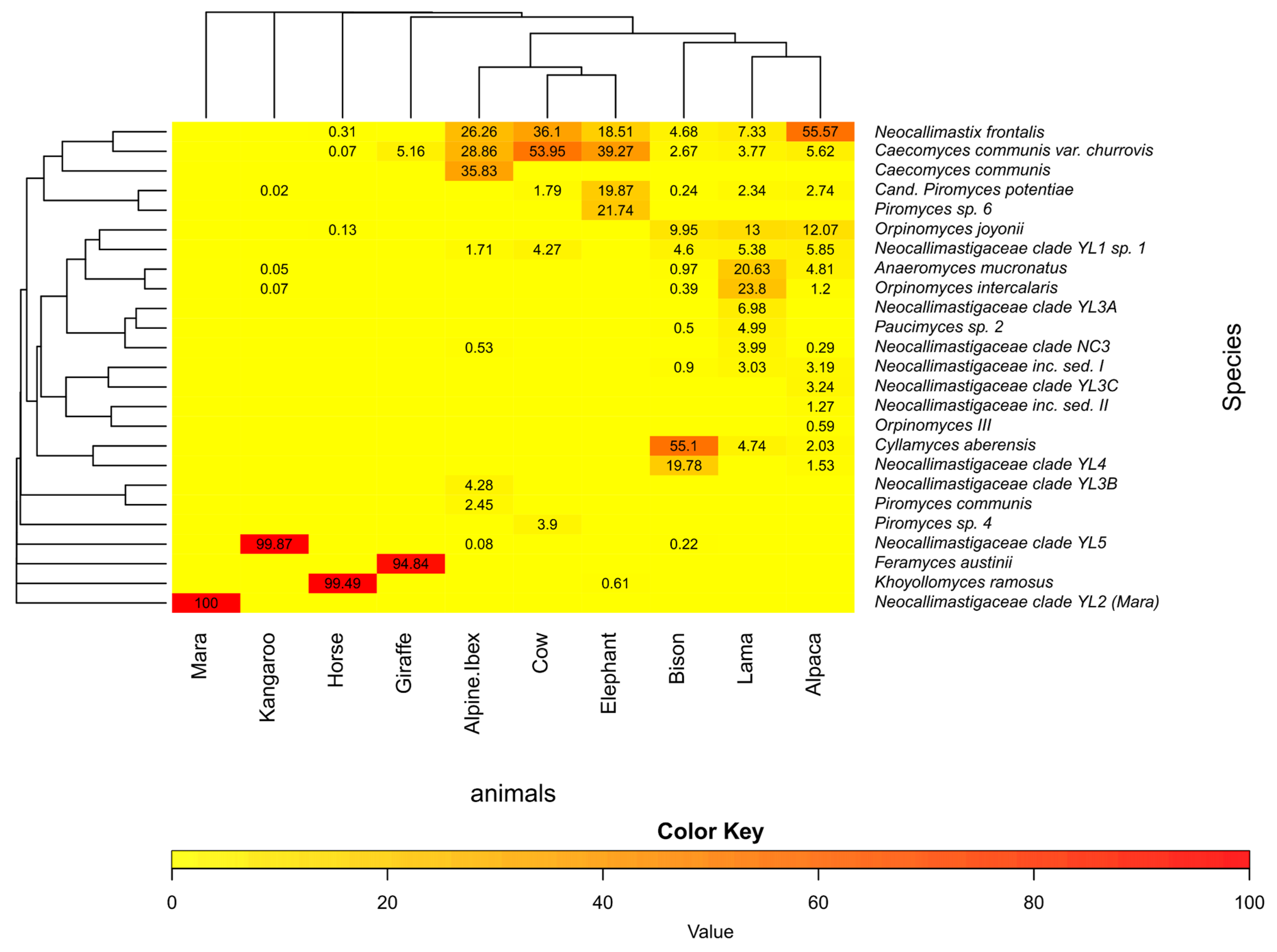
3.5. Community Analysis
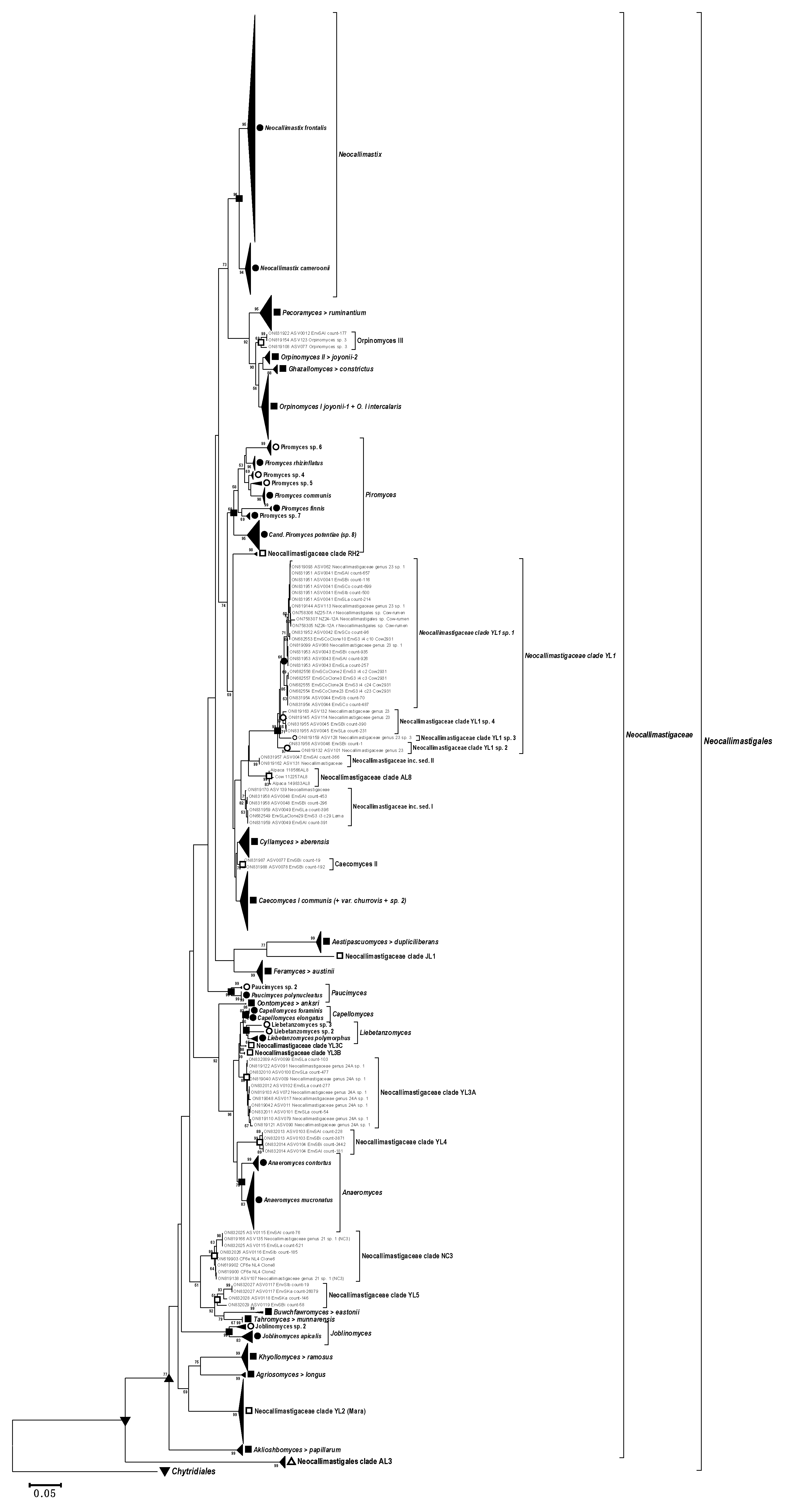
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanafy, R.A.; Lanjekar, V.B.; Dhakephalkar, P.K.; Callaghan, T.M.; Dagar, S.S.; Griffith, G.W.; Elshahed, M.S.; Youssef, N.H. Seven New Neocallimastigomycota Genera from Wild, Zoo-Housed, and Domesticated Herbivores Greatly Expand the Taxonomic Diversity of the Phylum. Mycologia 2020, 112, 1212–1239. [Google Scholar] [CrossRef] [PubMed]
- Liggenstoffer, A.S.; Youssef, N.H.; Couger, M.B.; Elshahed, M.S. Phylogenetic Diversity and Community Structure of Anaerobic Gut Fungi (Phylum Neocallimastigomycota) in Ruminant and Non-Ruminant Herbivores. ISME J. 2010, 4, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Dollhofer, V.; Callaghan, T.M.; Reitberger, S.; Lebuhn, M.; Benz, J.P. Isolation, Identification and Characterization of Lignocellulolytic Aerobic and Anaerobic Fungi in One- and Two-Phase Biogas Plants. Bioresour. Technol. 2018, 268, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, T.; Zebeli, Q. The Present Role and New Potentials of Anaerobic Fungi in Ruminant Nutrition. J. Fungi. 2021, 7, 200. [Google Scholar] [CrossRef] [PubMed]
- Saye, L.M.G.; Navaratna, T.A.; Chong, J.P.J.; O’Malley, M.A.; Theodorou, M.K.; Reilly, M. The Anaerobic Fungi: Challenges and Opportunities for Industrial Lignocellulosic Biofuel Production. Microorganisms 2021, 9, 694. [Google Scholar] [CrossRef]
- Solomon, K.V.; Haitjema, C.H.; Henske, J.K.; Gilmore, S.P.; Borges-Rivera, D.; Lipzen, A.; Brewer, H.M.; Purvine, S.O.; Wright, A.T.; Theodorou, M.K.; et al. Early-Branching Gut Fungi Possess a Large, Comprehensive Array of Biomass-Degrading Enzymes. Science 2016, 351, 1192–1195. [Google Scholar] [CrossRef]
- Dollhofer, V.; Callaghan, T.M.; Griffith, G.W.; Lebuhn, M.; Bauer, J. Presence and Transcriptional Activity of Anaerobic Fungi in Agricultural Biogas Plants. Bioresour. Technol. 2017, 235, 131–139. [Google Scholar] [CrossRef]
- Lockhart, R.J.; Van Dyke, M.I.; Beadle, I.R.; Humphreys, P.; McCarthy, A.J. Molecular Biological Detection of Anaerobic Gut Fungi ( Neocallimastigales ) from Landfill Sites. Appl. Environ. Microbiol. 2006, 72, 5659–5661. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List; Bolchacova, E.; et al. Nuclear Ribosomal Internal Transcribed Spacer (ITS) Region as a Universal DNA Barcode Marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS Variability in the Kingdom Fungi as Expressed in the International Sequence Databases and Its Implications for Molecular Species Identification. Evol. Bioinf. 2008, 4, EBO.S653. [Google Scholar] [CrossRef]
- Callaghan, T.M.; Podmirseg, S.M.; Hohlweck, D.; Edwards, J.E.; Puniya, A.K.; Dagar, S.S.; Griffith, G.W. Buwchfawromyces Eastonii Gen. Nov., Sp. Nov.: A New Anaerobic Fungus (Neocallimastigomycota) Isolated from Buffalo Faeces. MycoKeys 2015, 9, 11–28. [Google Scholar]
- Edwards, J.E.; Forster, R.J.; Callaghan, T.M.; Dollhofer, V.; Dagar, S.S.; Cheng, Y.; Chang, J.; Kittelmann, S.; Fliegerova, K.; Puniya, A.K.; et al. PCR and Omics Based Techniques to Study the Diversity, Ecology and Biology of Anaerobic Fungi: Insights, Challenges and Opportunities. Front. Microbiol. 2017, 8, 1657. [Google Scholar] [CrossRef] [PubMed]
- Dagar, S.S.; Kumar, S.; Mudgil, P.; Singh, R.; Puniya, A.K. D1/D2 Domain of Large-Subunit Ribosomal DNA for Differentiation of Orpinomyces spp. Appl. Environ. Microbiol. 2011, 77, 6722–6725. [Google Scholar] [CrossRef] [PubMed]
- Dollhofer, V.; Callaghan, T.M.; Dorn-In, S.; Bauer, J.; Lebuhn, M. Development of Three Specific PCR-Based Tools to Determine Quantity, Cellulolytic Transcriptional Activity and Phylogeny of Anaerobic Fungi. J. Microbiol. Methods 2016, 127, 28–40. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Johnson, B.; Youssef, N.H.; Elshahed, M.S. Assessing Anaerobic Gut Fungal Diversity in Herbivores Using D1/D2 Large Ribosomal Subunit Sequencing and Multi-year Isolation. Environ. Microbiol. 2020, 22, 3883–3908. [Google Scholar] [CrossRef]
- Hausner, G.; Inglis, G.D.; Yanke, L.J.; Kawchuk, L.M.; McAllister, T.A. Analysis of Restriction Fragment Length Polymorphisms in the Ribosomal DNA of a Selection of Anaerobic Chytrids. Can. J. Bot. 2000, 78, 917–927. [Google Scholar]
- Fliegerová, K.; Mrázek, J.; Voigt, K. Differentiation of Anaerobic Polycentric Fungi by RDNA PCR-RFLP. Folia Microbiol. 2006, 51, 273–277. [Google Scholar] [CrossRef]
- Nagler, M.; Podmirseg, S.M.; Griffith, G.W.; Insam, H.; Ascher-Jenull, J. The Use of Extracellular DNA as a Proxy for Specific Microbial Activity. Appl. Microbiol. Biotechnol. 2018, 102, 2885–2898. [Google Scholar] [CrossRef]
- Murphy, C.L.; Youssef, N.H.; Hanafy, R.A.; Couger, M.B.; Stajich, J.E.; Wang, Y.; Baker, K.; Dagar, S.S.; Griffith, G.W.; Farag, I.F.; et al. Horizontal Gene Transfer as an Indispensable Driver for Evolution of Neocallimastigomycota into a Distinct Gut-Dwelling Fungal Lineage. Appl. Environ. Microbiol. 2019, 85, e00988-19. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ashelford, K.E. PRIMROSE: A Computer Program for Generating and Estimating the Phylogenetic Range of 16S RRNA Oligonucleotide Probes and Primers in Conjunction with the RDP-II Database. Nucleic Acids Res. 2002, 30, 3481–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Stabel, M.; Hanafy, R.A.; Schweitzer, T.; Greif, M.; Aliyu, H.; Flad, V.; Young, D.; Lebuhn, M.; Elshahed, M.S.; Ochsenreither, K.; et al. Aestipascuomyces Dupliciliberans Gen. Nov, Sp. Nov., the First Cultured Representative of the Uncultured SK4 Clade from Aoudad Sheep and Alpaca. Microorganisms 2020, 8, 1734. [Google Scholar] [CrossRef]
- Joshi, A.; Young, D.; Huang, L.; Mosberger, L.; Munk, B.; Vinzelj, J.; Flad, V.; Sczyrba, A.; Griffith, G.W.; Podmirseg, S.; et al. Effect of Growth Media on the Diversity of Neocallimastigomycetes from Non-Rumen Habitats. Microorganisms 2022. submitted. [Google Scholar]
- Lowe, S.E.; Theodorou, M.K.; Trinci, A.P.J.; Hespell, R.B. Growth of Anaerobic Rumen Fungi on Defined and Semi-Defined Media Lacking Rumen Fluid. Microbiology 1985, 131, 2225–2229. [Google Scholar] [CrossRef]
- Marvin-Sikkema, F.D.; Richardson, A.J.; Stewart, C.S.; Gottschal, J.C.; Prins, R.A. Influence of Hydrogen-Consuming Bacteria on Cellulose Degradation by Anaerobic Fungi. Appl. Environ. Microbiol. 1990, 56, 3793–3797. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Whiteley, A.S.; O’Donnell, A.G.; Bailey, M.J. Rapid Method for Coextraction of DNA and RNA from Natural Environments for Analysis of Ribosomal DNA- and RRNA-Based Microbial Community Composition. Appl. Environ. Microbiol. 2000, 66, 5488–5491. [Google Scholar] [CrossRef]
- Lebuhn, M.; Derenkó, J.; Rademacher, A.; Helbig, S.; Munk, B.; Pechtl, A.; Stolze, Y.; Prowe, S.; Schwarz, W.; Schlüter, A.; et al. DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison. Bioengineering 2016, 3, 7. [Google Scholar] [CrossRef]
- Munk, B.; Guebitz, G.M.; Lebuhn, M. Influence of Nitrogen-Rich Substrates on Biogas Production and on the Methanogenic Community under Mesophilic and Thermophilic Conditions. Anaerobe 2017, 46, 146–154. [Google Scholar] [CrossRef]
- Reitmeier, S.; Kiessling, S.; Neuhaus, K.; Haller, D. Comparing Circadian Rhythmicity in the Human Gut Microbiome. STAR Protoc. 2020, 1, 100148. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.M.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package; R Package Version 2.6-2; R Core Team: Vienna, Austria, 2022. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data; R Package Version 3.1.3; R Core Team: Vienna, Austria, 2022. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Elshahed, M.; Radwa, H.; Yanfen, C.; Dagar, S.S.; Edwards, J.E.; Flad, V.; Kateřina, O.F.; Griffith, G.W.; Kittelmann, S.; Lebuhn, M.; et al. On the Characterization and Rank Assignment Criteria for the Anaerobic Gut Fungi (Neocallimastigomycota). IJSEM 2021, 72, 005449. [Google Scholar]
- Nilsson, R.H.; Sánchez-García, M.; Ryberg, M.K.; Abarenkov, K.; Wurzbacher, C.; Kristiansson, E. Read Quality-Based Trimming of the Distal Ends of Public Fungal DNA Sequences Is Nowhere near Satisfactory. MycoKeys 2017, 26, 13–24. [Google Scholar] [CrossRef]
- Mazars, G.-R.; Theillet, C. Direct Sequencing by Thermal Asymmetric PCR. In PCR Protocols; Humana Press: Totowa, NJ, USA, 2003; Volume 226, pp. 355–360. [Google Scholar]
- Teunissen, M.J.; Op den Camp, H.J.M.; Orpin, C.G.; Huis in ’t Veld, J.H.J.; Vogels, G.D. Comparison of Growth Characteristics of Anaerobic Fungi Isolated from Ruminant and Non-Ruminant Herbivores during Cultivation in a Defined Medium. J. Gen. Microbiol. 1991, 137, 1401–1408. [Google Scholar] [CrossRef]
- Gordon, G.L.R.; Phillips, M.W. The Role of Anaerobic Gut Fungi in Ruminants. Nutr. Res. Rev. 1998, 11, 133–168. [Google Scholar] [CrossRef] [PubMed]
- Mura, E.; Edwards, J.; Kittelmann, S.; Kaerger, K.; Voigt, K.; Mrázek, J.; Moniello, G.; Fliegerova, K. Anaerobic Fungal Communities Differ along the Horse Digestive Tract. Fungal Biol. 2019, 123, 240–246. [Google Scholar] [CrossRef]
- Fliegerová, K.; Mrázek, J.; Hoffmann, K.; Zábranská, J.; Voigt, K. Diversity of Anaerobic Fungi within Cow Manure Determined by ITS1 Analysis. Folia Microbiol. 2010, 55, 319–325. [Google Scholar] [CrossRef]
- Schmidt, J.M.; Henken, S.; Dowd, S.E.; McLaughlin, R.W. Analysis of the Microbial Diversity in the Fecal Material of Giraffes. Curr. Microbiol. 2018, 75, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative Microbiome Profiling Links Gut Community Variation to Microbial Load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Isolate | Source | Cultivation/Extraction Reference | |
|---|---|---|---|---|
| 1 | Aestipascuomyces dupliciliberans | A252 | KIT | [24] |
| 2 | Agriosomyces longus | MS-2 | OSU | [1] |
| 3 | Aklioshbomyces papillarum | WT4 | OSU | [1] |
| 4 | Anaeromyces contortus | G3C | OSU | [1] |
| 5 | Anaeromyces mucronatus | YoDo3 | LfL | this study |
| 6 | Buwchfawromyces eastonii | GE09 | DoLS | this study |
| 7 | Caecomyces communisvar. churrovis | ViSuPo1A | UIBK | this study |
| 8 | Caecomyces communisvar. churrovis | YoDo26 | LfL | this study |
| 9 | Capellomyces foraminis | BGB-12 | OSU | [1] |
| 10 | Feramyces austinii | DF1 | IAPG | this study |
| 11 | Ghazallomyces constrictus | ADS-11 | OSU | [1] |
| 12 | Khoyollomyces ramosus | H1WF70RF2 | ZHAW | [25] |
| 13 | Khoyollomyces ramosus | ZC-41 | OSU | [1] |
| 14 | Liebetanzomyces polymorphus | ORC-37 | OSU | [1] |
| 15 | Neocallimastix cameroonii | YoDo11 | LfL | this study |
| 16 | Neocallimastix frontalis | YoDo8 | LfL | this study |
| 17 | Orpinomyces cf. joyonii | D3B | OSU | [1] |
| 18 | Paucimyces polynucleatus | BB-3 | OSU | [1] |
| 19 | Pecoramyces ruminantium | OS2 | OSU | [1] |
| 20 | Piromyces communis | P | DoLS | this study |
| 21 | Chytridiomycetes sp. | AUS_17 | OL | outgroup, this study |
| Sample Source | AGF Host | Origin | Characteristics | Analysis | |
|---|---|---|---|---|---|
| 1 | Cow feces | Bos taurus Fleckvieh breed | Cattle farm in Dillingen, Germany | Ruminant with 4 compartments | (q)PCR, HTS, clone library |
| 2 | Lama feces | Lama glama | Munich Zoo, originally from South America | Ruminant with 3 compartments | (q)PCR, HTS, clone library |
| 3 | Horse feces | Equus ferus caballus | Private stable in Freising, Germany | Monogastric | (q)PCR, HTS |
| 4 | Alpine ibex feces | Capra ibex ibex | Munich zoo, originally from European alps | Ruminant with 4 compartments | (q)PCR, HTS |
| 5 | Alpaca feces | Vicugna pacos | Munich zoo, originally from South America | Ruminant with 3 compartments | (q)PCR, HTS |
| 6 | Kangaroo feces | Macropus rufus | Munich zoo, originally from Australia | Non-ruminant with 2 compartments | (q)PCR, HTS |
| 7 | Wood Bison feces | Bison bison athabascae | Munich zoo, originally from North America | Ruminant with 4 compartments | (q)PCR, HTS |
| 8 | Elephant feces | Elephas maximus | Munich zoo, originally from Asia | Monogastric | (q)PCR, HTS, clone library |
| 9 | Giraffe feces | Giraffa camelopardalis reticulata | Munich zoo, originally from East Africa | Ruminant with 4 compartments | (q)PCR, HTS |
| 10 | Mara feces | Dolichotis patagonum | Munich zoo, originally from Patagonia, South America | Monogastric, Rodent | (q)PCR, HTS, clone library |
| 11 | Pig feces | no known AGF host | Pig farm, Germany | Monogastric | (q)PCR |
| 12 | Dog feces | no known AGF host | Pet, Freising, Germany | Monogastric | (q)PCR |
| 13 | Digestate from fermenter of agricultural biogas plant PB 8 | Substrates from no AGF host | Operating biogas plant, Germany | Feedstock only of energy plants (triticale, grass, maize silage) | (q)PCR |
| 14 | Digestate from fermenter of agricultural biogas plant PB 25 | 36% of the substrates comes from an AGF host | Operating biogas plant, Germany | biogas plant fed with 36% cow manure | (q)PCR |
| 15 | Soil | no AGF host | Freising, Germany | Grassland, not of agricultural use | (q)PCR |
| Primer Name | Sequence (5′→3′) | Length (Bases) | Tm (°C) * | Amplicon Size (bp) | Target |
|---|---|---|---|---|---|
| AGF-LSU-EnvS F | GCGTTTRRCACCASTGTTGTT | 21 | 59.8 | 349–355 | D2 region of the 28S rRNA coding gene (LSU) |
| AGF-LSU-EnvS R | GTCAACATCCTAAGYGTAGGTA | 22 | 58.4 |
| Mock Community 1 | Mock Community 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Isolate | Reads per Isolate | % | % Pooled | Number of ASVs | Reads per Isolate | % | % Pooled | Number of ASVs |
| Aestipascuomyces dupliciliberans A252 | 254 | 3.2% | 5.3% | 4 | - | |||
| Agriosomyces longus MS-2 | 410 | 5.2% | 5.3% | 2 | 608 | 8.4% | 10.0% | 2 |
| Aklioshbomyces papillarum WT-4 | 371 | 4.7% | 5.3% | 2 | - | |||
| Anaeromyces contortus G3C | 394 | 5.0% | 5.3% | 5 | 8 | 0.1% | - | 1 |
| Anaeromyces mucronatus YoDo3 | 290 | 3.7% | 5.3% | 2 | - | |||
| Buwchfawromyces eastonii GE09 | 460 | 5.9% | 5.3% | 2 | - | |||
| Caecomyces communis var. churrovis ViSoPu1A | 837 | 10.7% | 10.5% | 2 | - | |||
| Capellomyces foraminis BGB-12 | 255 | 3.3% | 5.3% | 1 | - | |||
| Feramyces austinii DF1 | 391 | 5.0% | 5.3% | 3 | - | |||
| Ghazallomyces constrictus ADS-11 | 433 | 5.5% | 5.3% | 2 | - | |||
| Khoyollomyces ramosus H1WF70RF2 | 485 | 6.2% | 5.3% | 2 | 6560 | 90.8% | 90.0% | 2 |
| Liebetanzomyces polymorphus ORC-37 | 419 | 5.3% | 5.3% | 1 | - | |||
| Neocallimastix cameroonii YoDo11 | 448 | 5.7% | 5.3% | 4 | - | |||
| Neocallimastix frontalis YoDo8 | 515 | 6.6% | 5.3% | 4 | - | |||
| Orpinomyces cf. joyonii D3B | 449 | 5.7% | 5.3% | 4 | - | |||
| Paucimyces polynucleatus BB-3 | 537 | 6.9% | 5.3% | 1 | - | |||
| Pecoramyces ruminantium OS2 | 434 | 5.5% | 5.3% | 2 | - | |||
| Piromyces communis P | 455 | 5.8% | 5.3% | 3 * | 42 | 0.6% | - | 1 |
| Not assigned | 0 | 0.0% | - | 0 | 3 | 0.04% | - | 1 |
| Total | 7837 | 100% | 100% | 7221 | 100% | 100% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, D.; Joshi, A.; Huang, L.; Munk, B.; Wurzbacher, C.; Youssef, N.H.; Elshahed, M.S.; Moon, C.D.; Ochsenreither, K.; Griffith, G.W.; et al. Simultaneous Metabarcoding and Quantification of Neocallimastigomycetes from Environmental Samples: Insights into Community Composition and Novel Lineages. Microorganisms 2022, 10, 1749. https://doi.org/10.3390/microorganisms10091749
Young D, Joshi A, Huang L, Munk B, Wurzbacher C, Youssef NH, Elshahed MS, Moon CD, Ochsenreither K, Griffith GW, et al. Simultaneous Metabarcoding and Quantification of Neocallimastigomycetes from Environmental Samples: Insights into Community Composition and Novel Lineages. Microorganisms. 2022; 10(9):1749. https://doi.org/10.3390/microorganisms10091749
Chicago/Turabian StyleYoung, Diana, Akshay Joshi, Liren Huang, Bernhard Munk, Christian Wurzbacher, Noha H. Youssef, Mostafa S. Elshahed, Christina D. Moon, Katrin Ochsenreither, Gareth W. Griffith, and et al. 2022. "Simultaneous Metabarcoding and Quantification of Neocallimastigomycetes from Environmental Samples: Insights into Community Composition and Novel Lineages" Microorganisms 10, no. 9: 1749. https://doi.org/10.3390/microorganisms10091749
APA StyleYoung, D., Joshi, A., Huang, L., Munk, B., Wurzbacher, C., Youssef, N. H., Elshahed, M. S., Moon, C. D., Ochsenreither, K., Griffith, G. W., Callaghan, T. M., Sczyrba, A., Lebuhn, M., & Flad, V. (2022). Simultaneous Metabarcoding and Quantification of Neocallimastigomycetes from Environmental Samples: Insights into Community Composition and Novel Lineages. Microorganisms, 10(9), 1749. https://doi.org/10.3390/microorganisms10091749