
A Pipeline for Constructing Reference Genomes for Large Cohort-Specific Metagenome Compression

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohort Description
2.2. Data Pre-Processing and Taxonomic Profiling
2.3. Data Analysis
2.4. Construction of the Basic Reference Database
2.5. Evaluation of Short-Read Sequence Aligners
2.6. Construction of Cohort-Specific Reference Genomes and Compression
3. Results
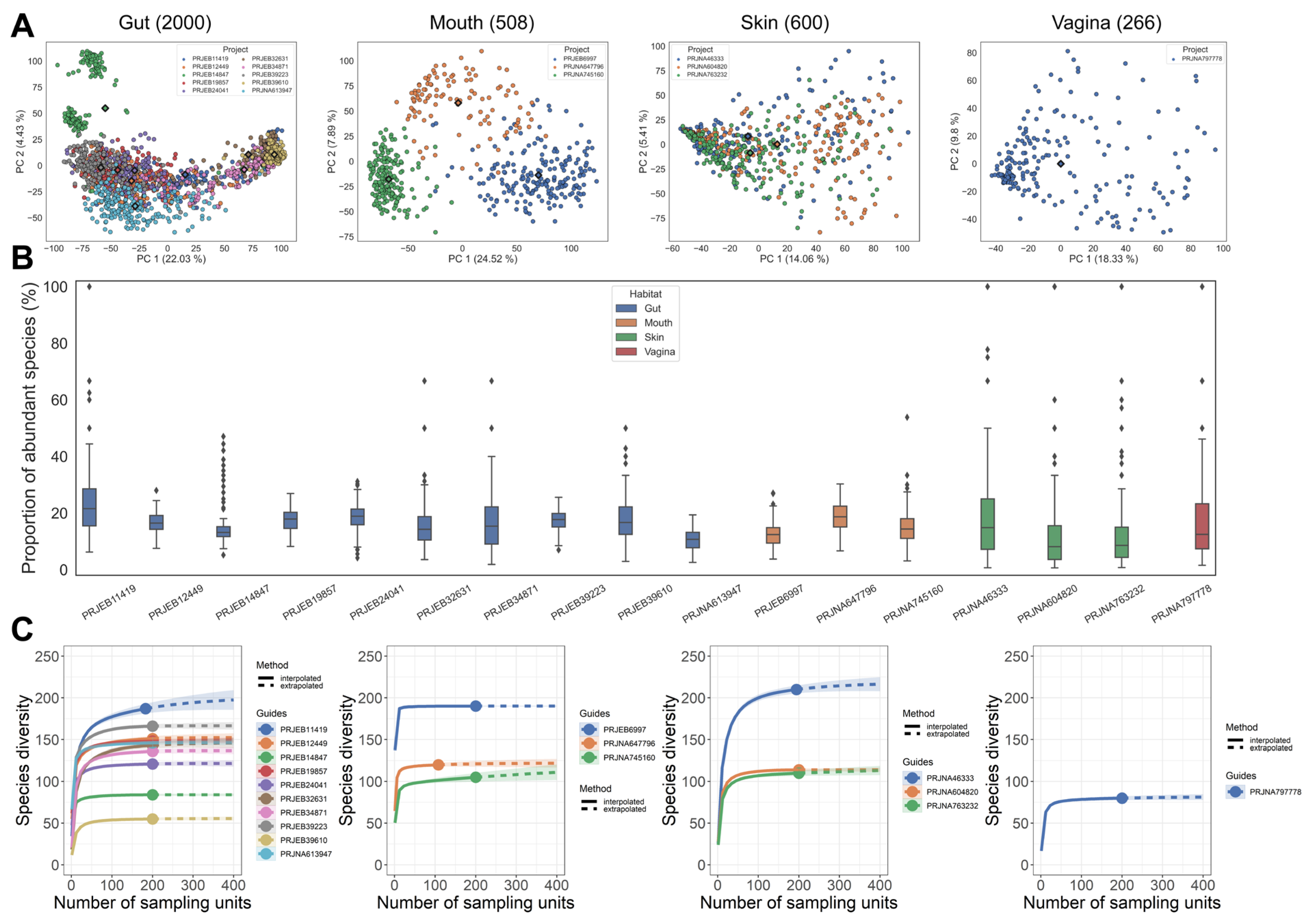
3.1. Microbiota Composition Analysis of Samples from Various Cohorts
3.2. Workflow of Constructing Reference Genomes for Specific Cohorts
3.3. Evaluation of Short-Read Sequence Aligners
3.4. Construction of Specific Reference Genomes for Each Dataset
3.5. Performance of Reference Genome-Based Compression of Each Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methe, B.A.; Huttenhower, C. The Human Microbiome Project: A community resource for the healthy human microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.D.; Consortium, M. MetaHIT: The European Union Project on metagenomics of the human intestinal tract. In Metagenomics of the Human Body; Nelson, K.E., Ed.; Springer: New York, NY, USA, 2011; pp. 307–316. ISBN 978-1-4419-7089-3. [Google Scholar]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Tara Oceans, C.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E.; et al. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Bonfield, J.K.; Mahoney, M.V. Compression of FASTQ and SAM format sequencing data. PLoS ONE 2013, 8, e59190. [Google Scholar] [CrossRef]
- Hach, F.; Numanagic, I.; Alkan, C.; Sahinalp, S.C. SCALCE: Boosting sequence compression algorithms using locally consistent encoding. Bioinformatics 2012, 28, 3051–3057. [Google Scholar] [CrossRef]
- Selva, J.J.; Chen, X. SRComp: Short read sequence compression using burstsort and Elias omega coding. PLoS ONE 2013, 8, e81414. [Google Scholar] [CrossRef]
- Janin, L.; Schulz-Trieglaff, O.; Cox, A.J. BEETL-fastq: A searchable compressed archive for DNA reads. Bioinformatics 2014, 30, 2796–2801. [Google Scholar] [CrossRef]
- Fritz, M.H.-Y.; Leinonen, R.; Cochrane, G.; Birney, E. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res. 2011, 21, 734–740. [Google Scholar] [CrossRef]
- Huang, Z.A.; Wen, Z.; Deng, Q.; Chu, Y.; Sun, Y.; Zhu, Z. LW-FQZip 2: A parallelized reference-based compression of FASTQ files. BMC Bioinform. 2017, 18, 179. [Google Scholar] [CrossRef]
- Jones, D.C.; Ruzzo, W.L.; Peng, X.; Katze, M.G. Compression of next-generation sequencing reads aided by highly efficient de novo assembly. Nucleic Acids Res. 2012, 40, e171. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Tobler, R.; Souilmi, Y.; Llamas, B. Genozip—A Universal Extensible Genomic Data Compressor. Bioinformatics 2021, 37, 2225–2230. [Google Scholar] [CrossRef] [PubMed]
- Hach, F.; Numanagic, I.; Sahinalp, S.C. DeeZ: Reference-based compression by local assembly. Nat. Methods 2014, 11, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015, 44, D733–D745. [Google Scholar] [CrossRef]
- Youens-Clark, K.; Bomhoff, M.; Ponsero, A.J.; Wood-Charlson, E.M.; Lynch, J.; Choi, I.; Hartman, J.H.; Hurwitz, B.L. iMicrobe: Tools and data-dreaiven discovery platform for the microbiome sciences. GigaScience 2019, 8, giz083. [Google Scholar] [CrossRef]
- Mitchell, A.; Bucchini, F.; Cochrane, G.; Denise, H.; ten Hoopen, P.; Fraser, M.; Pesseat, S.; Potter, S.; Scheremetjew, M.; Sterk, P.; et al. EBI metagenomics in 2016–An expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Res. 2016, 44, D595–D603. [Google Scholar] [CrossRef]
- Chen, I.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef]
- Vogtmann, E.; Hua, X.; Zeller, G.; Sunagawa, S.; Voigt, A.Y.; Hercog, R.; Goedert, J.J.; Shi, J.; Bork, P.; Sinha, R. Colorectal Cancer and the Human Gut Microbiome: Reproducibility with Whole-Genome Shotgun Sequencing. PLoS ONE 2016, 11, e0155362. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJEB19857). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJEB19857 (accessed on 1 August 2022).
- Korpela, K.; Costea, P.; Coelho, L.P.; Kandels-Lewis, S.; Willemsen, G.; Boomsma, D.I.; Segata, N.; Bork, P. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 2018, 28, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Forster, S.C.; Tsaliki, E.; Vervier, K.; Strang, A.; Simpson, N.; Kumar, N.; Stares, M.D.; Rodger, A.; Brocklehurst, P.; et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 2019, 574, 117–121. [Google Scholar] [CrossRef]
- Auguet, O.T.; Niehus, R.; Gweon, H.S.; Berkley, J.A.; Waichungo, J.; Njim, T.; Edgeworth, J.D.; Batra, R.; Chau, K.; Swann, J.; et al. Population-level faecal metagenomic profiling as a tool to predict antimicrobial resistance in Enterobacterales isolates causing invasive infections: An exploratory study across Cambodia, Kenya, and the UK. E Clin. Med. 2021, 36, 100910. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Berry, S.E.; Valdes, A.M.; Nguyen, L.H.; Piccinno, G.; Drew, D.A.; Leeming, E.; Gibson, R.; Le Roy, C.; Khatib, H.A.; et al. Microbiome connections with host metabolism and habitual diet from 1098 deeply phenotyped individuals. Nat. Med. 2021, 27, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.C.; Embleton, N.D.; Lamb, C.A.; Young, G.; Granger, C.L.; Najera, J.; Smith, D.P.; Hoffman, K.L.; Petrosino, J.F.; Bode, L.; et al. Human milk oligosaccharide DSLNT and gut microbiome in preterm infants predicts necrotising enterocolitis. Gut 2021, 70, 2273–2282. [Google Scholar] [CrossRef]
- Valles-Colomer, M.; Blanco-Míguez, A.; Manghi, P.; Asnicar, F.; Dubois, L.; Golzato, D.; Armanini, F.; Cumbo, F.; Huang, K.D.; Manara, S.; et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature 2023, 614, 125–135. [Google Scholar] [CrossRef]
- Pettigrew, M.M.; Kwon, J.; Gent, J.F.; Kong, Y.; Wade, M.; Williams, D.J.; Creech, C.B.; Evans, S.; Pan, Q.; Walter, E.B.; et al. Comparison of the Respiratory Resistomes and Microbiota in Children Receiving Short versus Standard Course Treatment for Community-Acquired Pneumonia. mBio 2022, 13, e0019522. [Google Scholar] [CrossRef]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJNA647796). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJNA647796 (accessed on 1 August 2022).
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef]
- Jo, J.H.; Harkins, C.P.; Schwardt, N.H.; Portillo, J.A.; Zimmerman, M.D.; Carter, C.L.; Hossen, M.A.; Peer, C.J.; Polley, E.C.; Dartois, V.; et al. Alterations of human skin microbiome and expansion of antimicrobial resistance after systemic antibiotics. Sci. Transl. Med. 2021, 13, eabd8077. [Google Scholar] [CrossRef] [PubMed]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJNA763232). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJNA763232 (accessed on 1 August 2022).
- France, M.T.; Fu, L.; Rutt, L.; Yang, H.; Humphrys, M.S.; Narina, S.; Gajer, P.M.; Ma, B.; Forney, L.J.; Ravel, J. Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data. Genome Biol. 2022, 23, 66. [Google Scholar] [CrossRef] [PubMed]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJEB42019). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJEB42019 (accessed on 1 August 2022).
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJEB24121). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJEB24121 (accessed on 1 August 2022).
- Bahram, M.; Espenberg, M.; Pärn, J.; Lehtovirta-Morley, L.; Anslan, S.; Kasak, K.; Kõljalg, U.; Liira, J.; Maddison, M.; Moora, M.; et al. Structure and function of the soil microbiome underlying N2O emissions from global wetlands. Nat. Commun. 2022, 13, 1430. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.A.; Garcia, C.A.; Garcia, N.; Brock, M.L.; Lee, J.A.; Ustick, L.J.; Barbero, L.; Carter, B.R.; Sonnerup, R.E.; Talley, L.D.; et al. High spatial resolution global ocean metagenomes from Bio-GO-SHIP repeat hydrography transects. Sci. Data 2021, 8, 107. [Google Scholar] [CrossRef]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJNA681031). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJNA681031 (accessed on 1 August 2022).
- Schultz, D. Mechanisms of Polysaccharide-Degradation in Particle-Associated Microbial Communities. Available online: https://nbn-resolving.org/urn:nbn:de:gbv:9-opus-59014 (accessed on 1 August 2022).
- Rossum, T.V.; Uyaguari-Diaz, M.I.; Vlok, M.; Peabody, M.A.; Tian, A.; Cronin, K.I.; Chan, M.; Croxen, M.A.; Hsiao, W.W.L.; Isaac-Renton, J.; et al. Spatiotemporal dynamics of river viruses, bacteria and microeukaryotes. bioRxiv 2018, 259861. [Google Scholar] [CrossRef]
- Pérez-Carrascal, O.M.; Tromas, N.; Terrat, Y.; Moreno, E.; Giani, A.; Corrêa Braga Marques, L.; Fortin, N.; Shapiro, B.J. Single-colony sequencing reveals microbe-by-microbiome phylosymbiosis between the cyanobacterium Microcystis and its associated bacteria. Microbiome 2021, 9, 194. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, Q.; Liao, K.; Jian, Z.; Zhao, C.; Qu, J. Fungal Community as a Bioindicator to Reflect Anthropogenic Activities in a River Ecosystem. Front. Microbiol. 2018, 9, 3152. [Google Scholar] [CrossRef]
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJNA746354). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJNA746354 (accessed on 1 August 2022).
- Pre-BreedYield Consortium EMBL Nucleotide Sequence Database (Project PRJNA801677). Available online: https://www.ebi.ac.uk/ena/browser/view/PRJNA801677 (accessed on 1 August 2022).
- Poulsen, C.S.; Ekstrom, C.T.; Aarestrup, F.M.; Pamp, S.J. Library Preparation and Sequencing Platform Introduce Bias in Metagenomic-Based Characterizations of Microbiomes. Microbiol. Spectr. 2022, 10, e0009022. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Miguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (H ill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Avolio, M.L.; Forrestel, E.J.; Chang, C.C.; La Pierre, K.J.; Burghardt, K.T.; Smith, M.D. Demystifying dominant species. New Phytol. 2019, 223, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Loftus, M.; Hassouneh, S.A.; Yooseph, S. Bacterial associations in the healthy human gut microbiome across populations. Sci. Rep. 2021, 11, 2828. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jiang, X.; Wang, S.; Kim, J.; Xiong, H.; Ohno-Machado, L. HUGO: Hierarchical mUlti-reference Genome compression for aligned reads. J. Am. Med. Inform. Assoc. 2014, 21, 363–373. [Google Scholar] [CrossRef]
- Deorowicz, S.; Danek, A. GTShark: Genotype compression in large projects. Bioinformatics 2019, 35, 4791–4793. [Google Scholar] [CrossRef]
- Dufort y Álvarez, G.; Seroussi, G.; Smircich, P.; Sotelo-Silveira, J.; Ochoa, I.; Martín, Á. RENANO: A REference-based compressor for NANOpore FASTQ files. Bioinformatics 2021, 37, 4862–4864. [Google Scholar] [CrossRef]
- Ochoa, I.; Asnani, H.; Bharadia, D.; Chowdhury, M.; Weissman, T.; Yona, G. QualComp: A new lossy compressor for quality scores based on rate distortion theory. BMC Bioinform. 2013, 14, 187. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Zhu, J.; Tian, L.; Chen, P.; Han, M.; Song, L.; Tong, X.; Sun, X.; Yang, F.; Lin, Z.; Liu, X.; et al. Over 50,000 Metagenomically Assembled Draft Genomes for the Human Oral Microbiome Reveal New Taxa. Genom. Proteom. Bioinform. 2022, 20, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Yang, F.; Stepanauskas, R.; Cardenas, E.; Garoutte, A.; Williams, R.; Flater, J.; Tiedje, J.M.; Hofmockel, K.S.; Gelder, B.; et al. Strategies to improve reference databases for soil microbiomes. ISME J. 2017, 11, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Dunivin, T.K.; Choi, J.; Howe, A.; Shade, A. RefSoil+: A Reference Database for Genes and Traits of Soil Plasmids. mSystems 2019, 4, e00349-18. [Google Scholar] [CrossRef] [PubMed]
- Klemetsen, T.; Raknes, I.A.; Fu, J.; Agafonov, A.; Balasundaram, S.V.; Tartari, G.; Robertsen, E.; Willassen, N.P. The MAR databases: Development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2017, 46, D692–D699. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Habitats | BioProject | Sample Size | Data Size | Sample Used in This Study | References |
|---|---|---|---|---|---|
| Human gut | PRJEB11419 | 39,038 | 1003.18 Gb | 200 | [20] |
| PRJEB12449 | 882 | 371.38 Gb | 200 | [21] | |
| PRJEB14847 | 372 | 1.18 Tb | 200 | [22] | |
| PRJEB19857 | 350 | 520.69 Gb | 200 | [23] | |
| PRJEB24041 | 633 | 480.72 Gb | 200 | [24] | |
| PRJEB32631 | 1679 | 1.34 Tb | 200 | [25] | |
| PRJEB34871 | 1197 | 1.13 Tb | 200 | [26] | |
| PRJEB39223 | 2196 | 2.97 Tb | 200 | [27] | |
| PRJEB39610 | 644 | 974.51 Gb | 200 | [28] | |
| PRJNA613947 | 348 | 871.69 Gb | 200 | [29] | |
| Human mouth | PRJNA745160 | 888 | 657.43 Gb | 200 | [30] |
| PRJNA647796 | 108 | 219.63 Gb | 108 | [31] | |
| PRJEB6997 | 530 | 2.01 Tb | 200 | [32] | |
| Human skin | PRJNA46333 | 8774 | 1.87 Tb | 200 | [33] |
| PRJNA604820 | 516 | 269.29 Gb | 200 | [34] | |
| PRJNA763232 | 289 | 210.76 Gb | 200 | [35] | |
| Human vagina | PRJNA797778 | 542 | 649.33 Gb | 200 | [36] |
| Soil | PRJEB42019 | 7557 | 726.36 Gb | 200 | [37] |
| PRJEB24121 | 290 | 180.83 Gb | 200 | [38] | |
| PRJEB44414 | 195 | 239.80 Gb | 195 | [39] | |
| Marine | PRJNA656268 | 1942 | 1.16 Tb | 200 | [40] |
| PRJNA681031 | 305 | 268.79 Gb | 200 | [41] | |
| PRJEB38290 | 308 | 1.11 Tb | 200 | [42] | |
| Freshwater | PRJNA287840 | 729 | 154.65 Gb | 200 | [43] |
| PRJNA662092 | 184 | 265.07 Gb | 184 | [44] | |
| PRJNA418866 | 790 | 335.47 Gb | 116 | [45] | |
| Wastewater | PRJNA746354 | 3188 | 447.20 Gb | 200 | [46] |
| PRJNA801677 | 2105 | 452.38 Gb | 200 | [47] | |
| PRJEB31650 | 567 | 689.20 Gb | 200 | [48] |
| Aligner | Ref1000 (3.8 GB) | |
|---|---|---|
| Time (s) | Mem (GB) | |
| Bowtie2 | 15,081 | 10 |
| BWA | 5821 | 6 |
| Minimap2 | 164 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Ding, R.; He, S.; Wang, Q.; Zhou, Y. A Pipeline for Constructing Reference Genomes for Large Cohort-Specific Metagenome Compression. Microorganisms 2023, 11, 2560. https://doi.org/10.3390/microorganisms11102560
Wang L, Ding R, He S, Wang Q, Zhou Y. A Pipeline for Constructing Reference Genomes for Large Cohort-Specific Metagenome Compression. Microorganisms. 2023; 11(10):2560. https://doi.org/10.3390/microorganisms11102560
Chicago/Turabian StyleWang, Linqi, Renpeng Ding, Shixu He, Qinyu Wang, and Yan Zhou. 2023. "A Pipeline for Constructing Reference Genomes for Large Cohort-Specific Metagenome Compression" Microorganisms 11, no. 10: 2560. https://doi.org/10.3390/microorganisms11102560