
Contamination of Plant Foods with Bacillus cereus in a Province and Analysis of Its Traceability



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Isolation and Identification
2.3. Whole-Genome Sequencing and Genome Assembly
2.3.1. DNA Library Construction
2.3.2. On-Board Sequencing
2.3.3. Raw Data Processing
2.3.4. Genome Assembly
2.4. Genome-Wide Bioinformatics Analysis
2.4.1. Subspecies Typing
2.4.2. MLST
2.4.3. Toxicity Gene Analysis
2.4.4. cgSNP Phylogenetic Analysis
2.4.5. Establishment of a Molecular Traceability System for B. cereus
3. Results
3.1. Isolation and Identification of B. cereus
3.2. Detection of B. cereus in Samples
3.2.1. Results of B. cereus Detection in Different Regions
3.2.2. Detection of Different Types of B. cereus
3.2.3. Detection of B. cereus in Different Sites
3.3. Statistics and Assembly of Sequencing Results
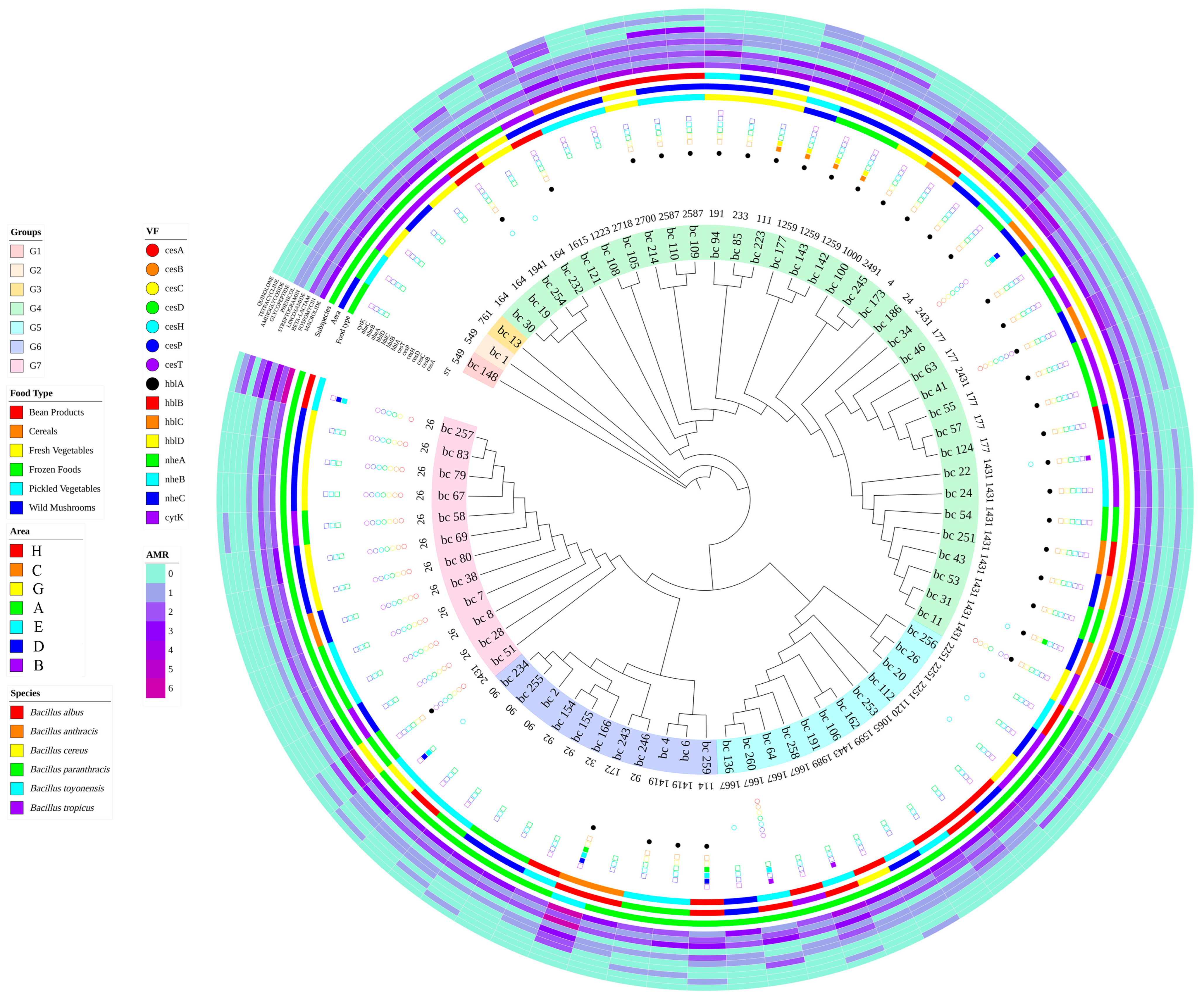
3.4. Molecular Typing of B. cereus
3.4.1. Results of B. cereus Subspecies Typing
3.4.2. MLST Results
3.4.3. MLST Results of B. cereus in Different Regions
3.4.4. MLST Results of B. cereus in Different Foodstuffs
3.5. Toxicity Gene Detection Results
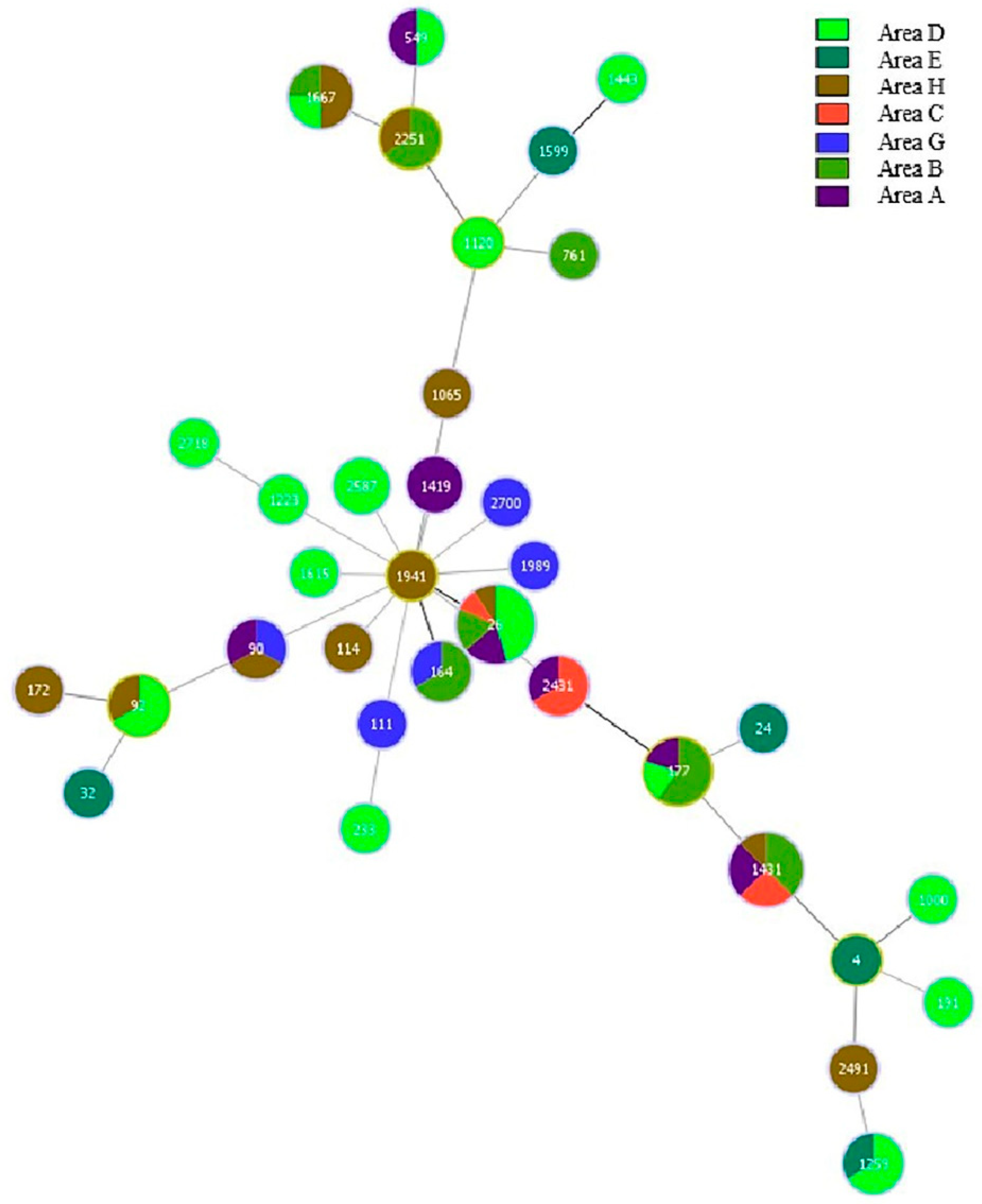
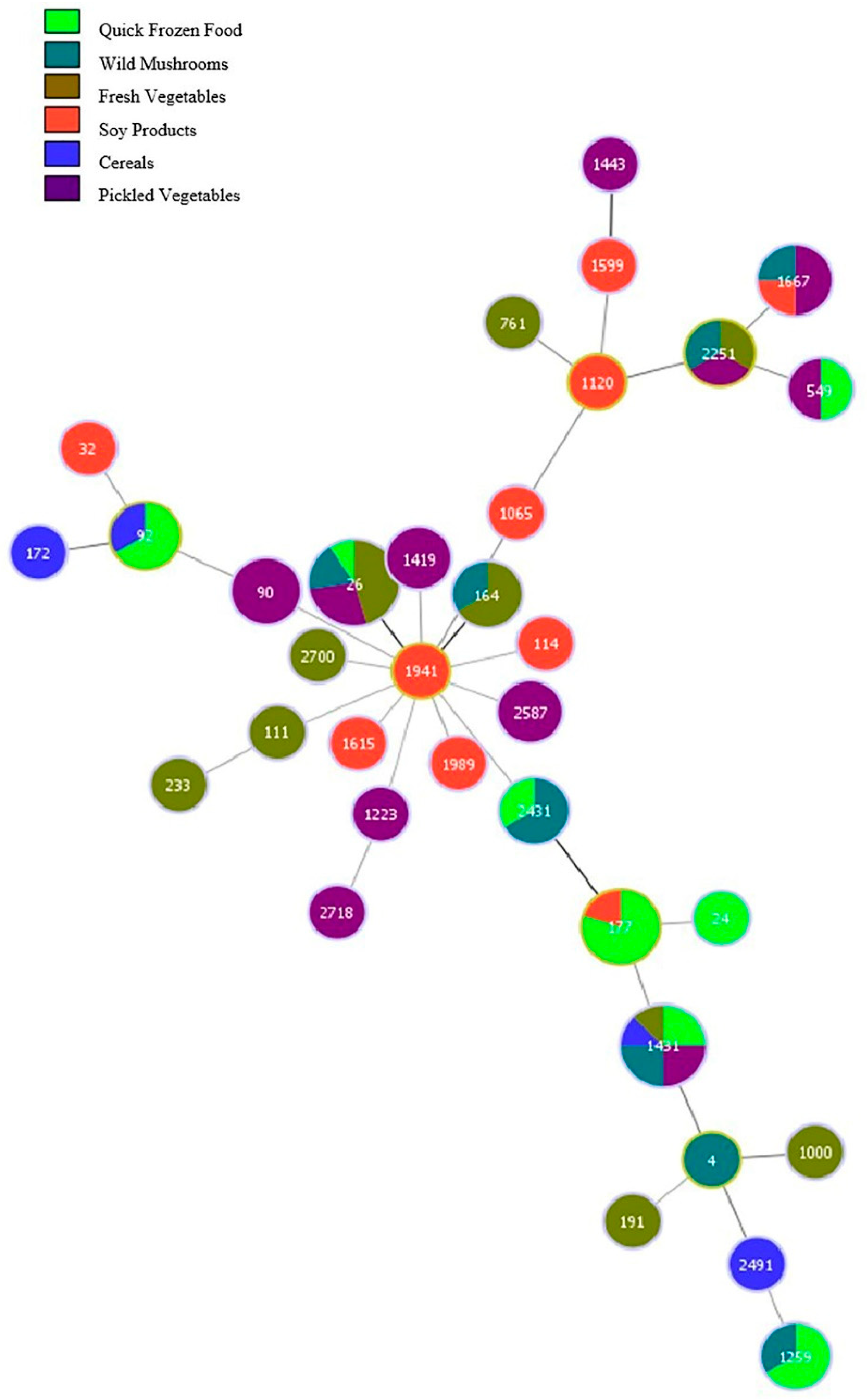
3.6. cgSNP Phylogenetic Analysis
3.7. Establishment of a Traceability System for B. cereus
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Terranova, W.; Blake, P.A. Bacillus Cereus Food Poisoning. N. Engl. J. Med. 1978, 298, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Bacillus Cereus, a Volatile Human Pathogen. Clin. Microbiol. Rev. 2010, 23, 382. [Google Scholar] [CrossRef] [PubMed]
- Drobniewski, F.A. Bacillus Cereus and Related Species. Clin. Microbiol. Rev. 1993, 6, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Lotte, R.; Chevalier, A.; Boyer, L.; Ruimy, R. Bacillus Cereus Invasive Infections in Preterm Neonates: An Up-to-Date Review of the Literature. Clin. Microbiol. Rev. 2022, 35, e00088-e21. [Google Scholar] [CrossRef]
- Yang, Y.; Gu, H.; Yu, X.; Zhan, L.; Chen, J.; Luo, Y.; Zhang, Y.; Zhang, Y.; Lu, Y.; Jiang, J.; et al. Genotypic Heterogeneity of Emetic Toxin Producing Bacillus Cereus Isolates from China. FEMS Microbiol. Lett. 2017, 364, fnw237. [Google Scholar] [CrossRef]
- Carter, L.; Huang, M.-C.J.; Han, K.; Gangiredla, J.; Yee, J.; Chase, H.R.; Negrete, F.; Tall, B.D. Characterization and Genetic Diversity of Bacillus Cereus Strains Isolated from Baby Wipes. Microorganisms 2022, 10, 1779. [Google Scholar] [CrossRef]
- Arnesen, L.P.S.; Fagerlund, A.; Granum, P.E. From Soil to Gut: Bacillus Cereus and Its Food Poisoning Toxins. Fems. Microbiol. Rev. 2008, 32, 579–606. [Google Scholar] [CrossRef]
- Tirloni, E.; Bernardi, C.; Celandroni, F.; Mazzantini, D.; Massimino, M.; Stella, S.; Ghelardi, E. Prevalence, Virulence Potential, and Growth in Cheese of Bacillus Cereus Strains Isolated from Fresh and Short-Ripened Cheeses Sold on the Italian Market. Microorganisms 2023, 11, 521. [Google Scholar] [CrossRef]
- Madoroba, E.; Magwedere, K.; Chaora, N.S.; Matle, I.; Muchadeyi, F.; Mathole, M.A.; Pierneef, R. Microbial Communities of Meat and Meat Products: An Exploratory Analysis of the Product Quality and Safety at Selected Enterprises in South Africa. Microorganisms 2021, 9, 507. [Google Scholar] [CrossRef]
- European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC). The European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17, 5926. [Google Scholar] [CrossRef]
- Messelhaeusser, U.; Frenzel, E.; Bloechinger, C.; Zucker, R.; Kaempf, P.; Ehling-Schulz, A. Emetic Bacillus Cereus Are More Volatile Than Thought: Recent Foodborne Outbreaks and Prevalence Studies in Bavaria (2007–2013). Biomed. Res. Int. 2014, 2014, 465603. [Google Scholar] [CrossRef]
- Callegan, M.C.; Kane, S.T.; Cochran, D.C.; Gilmore, M.S. Molecular Mechanisms of Bacillus Endophthalmitis Pathogenesis. DNA Cell Biol. 2002, 21, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.; Schrodt, C.A.; Feldmann, K.; Traxler, R.M.; Gee, J.E.; Kolton, C.B.; Marston, C.K.; Gulvik, C.A.; Antonini, J.M.; Negron, M.E.; et al. Fatal Anthrax Pneumonia in Welders and Other Metalworkers Caused by Bacillus Cereus Group Bacteria Containing Anthrax Toxin Genes-US Gulf Coast States, 1994–2020. MMWR-Morb. Mortal. Wkly. Rep. 2021, 70, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.K.; Lyman, J.A. Updated Review of Blood Culture Contamination. Clin. Microbiol. Rev. 2006, 19, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Glil, M.Y.; Chiaverini, A.; Garofolo, G.; Fasanella, A.; Parisi, A.; Harmsen, D.; Jolley, K.A.; Elschner, M.C.; Tomaso, H.; Linde, J.; et al. A Whole-Genome-Based Gene-by-Gene Typing System for Standardized High-Resolution Strain Typing of Bacillus Anthracis. J. Clin. Microbiol. 2021, 59, e02889-e20. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Beno, S.M.; Kent, D.J.; Carroll, L.M.; Martin, N.H.; Boor, K.J.; Kovac, J. Bacillus Wiedmannii Sp Nov., a Psychrotolerant and Cytotoxic Bacillus Cereus Group Species Isolated from Dairy Foods and Dairy Environments. Int. J. Syst. Evol. Microbiol. 2016, 66, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Kaptchouang Tchatchouang, C.-D.; Fri, J.; De Santi, M.; Brandi, G.; Schiavano, G.F.; Amagliani, G.; Ateba, C.N. Listeriosis Outbreak in South Africa: A Comparative Analysis with Previously Reported Cases Worldwide. Microorganisms 2020, 8, 135. [Google Scholar] [CrossRef]
- Perez-Losada, M.; Arenas, M.; Castro-Nallar, E. Microbial Sequence Typing in the Genomic Era. Infect. Genet. Evol. 2018, 63, 346–359. [Google Scholar] [CrossRef]
- Carroll, L.M.; Cheng, R.A.; Wiedmann, M.; Kovac, J. Keeping up with the Bacillus Cereus Group: Taxonomy through the Genomics Era and Beyond. Crit. Rev. Food Sci. Nutr. 2022, 62, 7677–7702. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, Y.M.; Li, L.; Yi, L.Z.; Zhao, W.W.; Shang, Y.; Cao, J.X. Advances in1 Traceability Typing and Identification of Foodborne Pathogens. Shipin Gongye Keji 2022, 43, 427–437. [Google Scholar] [CrossRef]
- Zervas, A.; Aggerbeck, M.R.; Allaga, H.; Güzel, M.; Hendriks, M.; Jonuškienė, I.; Kedves, O.; Kupeli, A.; Lamovšek, J.; Mülner, P.; et al. Identification and Characterization of 33 Bacillus Cereus Sensu Lato Isolates from Agricultural Fields from Eleven Widely Distributed Countries by Whole Genome Sequencing. Microorganisms 2020, 8, 2028. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; SenGupta, D.J.; Cummings, L.A.; Land, T.A.; Hoogestraat, D.R.; Cookson, B.T. Application of Whole-Genome Sequencing for Bacterial Strain Typing in Molecular Epidemiology. J. Clin. Microbiol. 2015, 53, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Apruzzese, I.; Song, E.; Bonah, E.; Sanidad, V.S.; Leekitcharoenphon, P.; Medardus, J.J.; Abdalla, N.; Hosseini, H.; Takeuchi, M. Investing in Food Safety for Developing Countries: Opportunities and Challenges in Applying Whole-Genome Sequencing for Food Safety Management. Foodborne Pathog. Dis. 2019, 16, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Pankhurst, L.J.; del Ojo Elias, C.; Votintseva, A.A.; Walker, T.M.; Cole, K.; Davies, J.; Fermont, J.M.; Gascoyne-Binzi, D.M.; Kohl, T.A.; Kong, C.; et al. Rapid, Comprehensive, and Affordable Mycobacterial Diagnosis with Whole-Genome Sequencing: A Prospective Study. Lancet Respir. Med. 2016, 4, 49–58. [Google Scholar] [CrossRef]
- Fox, E.M.; Casey, A.; Jordan, K.; Coffey, A.; Gahan, C.G.M.; McAuliffe, O. Whole Genome Sequence Analysis; an Improved Technology That Identifies Underlying Genotypic Differences between Closely Related Listeria Monocytogenes Strains. Innov. Food Sci. Emerg. Technol. 2017, 44, 89–96. [Google Scholar] [CrossRef]
- NIHR Global Health Research Unit on Genomic Surveillance of AMR. Whole-Genome Sequencing as Part of National and International Surveillance Programmes for Antimicrobial Resistance: A Roadmap. BMJ Glob. Health 2020, 5, e002244. [Google Scholar] [CrossRef]
- Revez, J.; Espinosa, L.; Albiger, B.; Leitmeyer, K.C.; Struelens, M.J.; ECDC National Microbiology Focal Points and Experts Group. Survey on the Use of Whole-Genome Sequencing for Infectious Diseases Surveillance: Rapid Expansion of European National Capacities, 2015–2016. Front. Public Health 2017, 5, 347. [Google Scholar] [CrossRef]
- GB 4789.14-2014; National Safety Standard for Food-Microbiological Examination of Food-Test for Bacillus cereus. China Standards Press: Beijing, China, 2015.
- Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Res 2018, 7, 1338. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Yang, J.L.; Zhang, Y.; Huang, K.N.; He, H.Y. Investigation of contaminated Bacillus cereus in sufu sold in Guizhou. J. Food Saf. Qual. 2021, 12, 2876–2880. [Google Scholar] [CrossRef]
- Du, Q.; Zhang, L.J.; Xu, X.Y. Investigation on polluting condition of food-borne pathogens in Changzhou from 2010 to 2012. Jiangsu J. Prev. Med. 2013, 24, 11–13. [Google Scholar]
- La, T.; Phillips, N.D.; Harland, B.L.; Wanchanthuek, P.; Bellgard, M.I.; Hampson, D.J. Multilocus Sequence Typing as a Tool for Studying the Molecular Epidemiology and Population Structure of Brachyspira Hyodysenteriae. Vet. Microbiol. 2009, 138, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Molecular Characteristics and Antibiotic Resistance of Bacillus cereus from Foods Using Whole Genome Sequencing-All Databases. Available online: https://www.webofscience.com/wos/alldb/full-record/CSCD:7078931 (accessed on 21 October 2023).
- Yang, Y.; Yu, X.; Zhan, L.; Chen, J.; Zhang, Y.; Zhang, J.; Chen, H.; Zhang, Z.; Zhang, Y.; Lu, Y.; et al. Multilocus Sequence Type Profiles of Bacillus Cereus Isolates from Infant Formula in China. Food Microbiol. 2017, 62, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, H.; Qiu, Z.; Jin, M.; Yang, D.; Xu, Q.; Feng, H.; Li, J.; Shen, Z. Identifying Geographic Origins of the Escherichia Coli Isolates from Food by a Method Based on Single-Nucleotide Polymorphisms. J. Microbiol. Methods 2020, 168, 105807. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, Y.; Carleton, H.; Timme, R.; Melka, D.; Muruvanda, T.; Wang, C.; Kastanis, G.; Katz, L.S.; Turner, L.; et al. Whole Genome and Core Genome Multilocus Sequence Typing and Single Nucleotide Polymorphism Analyses of Listeria Monocytogenes Isolates Associated with an Outbreak Linked to Cheese, United States, 2013. Appl. Environ. Microbiol. 2017, 83, e00633-e17. [Google Scholar] [CrossRef]
- Liu, Y.; Du, J.; Lai, Q.; Zeng, R.; Ye, D.; Xu, J.; Shao, Z. Proposal of Nine Novel Species of the Bacillus Cereus Group. Int. J. Syst. Evol. Microbiol. 2017, 67, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, L.P. Genomic and Pathogenicity of a Bacillus Paranthracis Isolated from Book Page Surface. Infect. Genet. Evol. 2021, 92, 104867. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, D.; Nolasco-Hipolito, C. Draft Genome Sequence of Bacillus Paranthracis Strain DB-4, Isolated from Nukadoko, Fermented Rice Bran for Japanese Pickles. Microbiol. Resour. Announc. 2021, 10, e00705–e00721. [Google Scholar] [CrossRef]
- Park, K.M.; Kim, H.J.; Jeong, M.C.; Koo, M. Occurrence of Toxigenic Bacillus Cereus and Bacillus Thuringiensis in Doenjang, a Korean Fermented Soybean Paste. J. Food Prot. 2016, 79, 605–612. [Google Scholar] [CrossRef]
- Okutani, A.; Inoue, S.; Noguchi, A.; Kaku, Y.; Morikawa, S. Whole-Genome Sequence-Based Comparison and Profiling of Virulence-Associated Genes of Bacillus Cereus Group Isolates from Diverse Sources in Japan. BMC Microbiol. 2019, 19, 296. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Wild Mushrooms | Soybean Products | Fresh Vegetables | Pickled Vegetables | Quick-Freeze Food | Cereals | Total |
|---|---|---|---|---|---|---|---|
| Area A | — | 1 | 1 | 9 | 3 | — | 14 |
| Area B | 3 | 5 | 11 | 10 | 4 | — | 33 |
| Area C | 12 | — | — | — | — | — | 12 |
| Area D | 10 | 8 | 15 | 11 | 14 | — | 58 |
| Area E | 5 | 14 | 22 | 4 | 4 | — | 49 |
| Area F | 2 | 19 | 19 | 6 | 7 | — | 53 |
| Area G | — | 24 | 12 | 4 | 2 | — | 42 |
| Area H | — | 5 | — | 3 | — | 4 | 12 |
| Total | 32 | 76 | 80 | 47 | 34 | 4 | 273 |
| Samples | Wild Mushrooms | Soybean Products | Fresh Vegetables | Pickled Vegetables | Quick-Freeze Food | Cereals | Total | Pollution Rate (%) |
|---|---|---|---|---|---|---|---|---|
| Area A | — | 1 | 0 | 3 | 3 | — | 6/14 | 42.86 |
| Area B | 2 | 5 | 3 | 1 | 3 | — | 10/33 | 30.30 |
| Area C | 5 | — | — | — | — | — | 5/12 | 41.67 |
| Area D | 1 | 3 | 6 | 3 | 3 | — | 16//58 | 27.59 |
| Area E | 2 | 2 | 0 | 0 | 1 | — | 5/49 | 10.20 |
| Area F | 0 | 0 | 0 | 0 | 0 | — | 0/53 | 0.00 |
| Area G | — | 1 | 3 | 1 | 0 | — | 5/42 | 11.90 |
| Area H | — | 2 | — | 3 | — | 4 | 9/12 | 75.00 |
| Total | 10/32 | 9/76 | 12/80 | 11/47 | 10/34 | 4/4 | 56/273 | 20.51 |
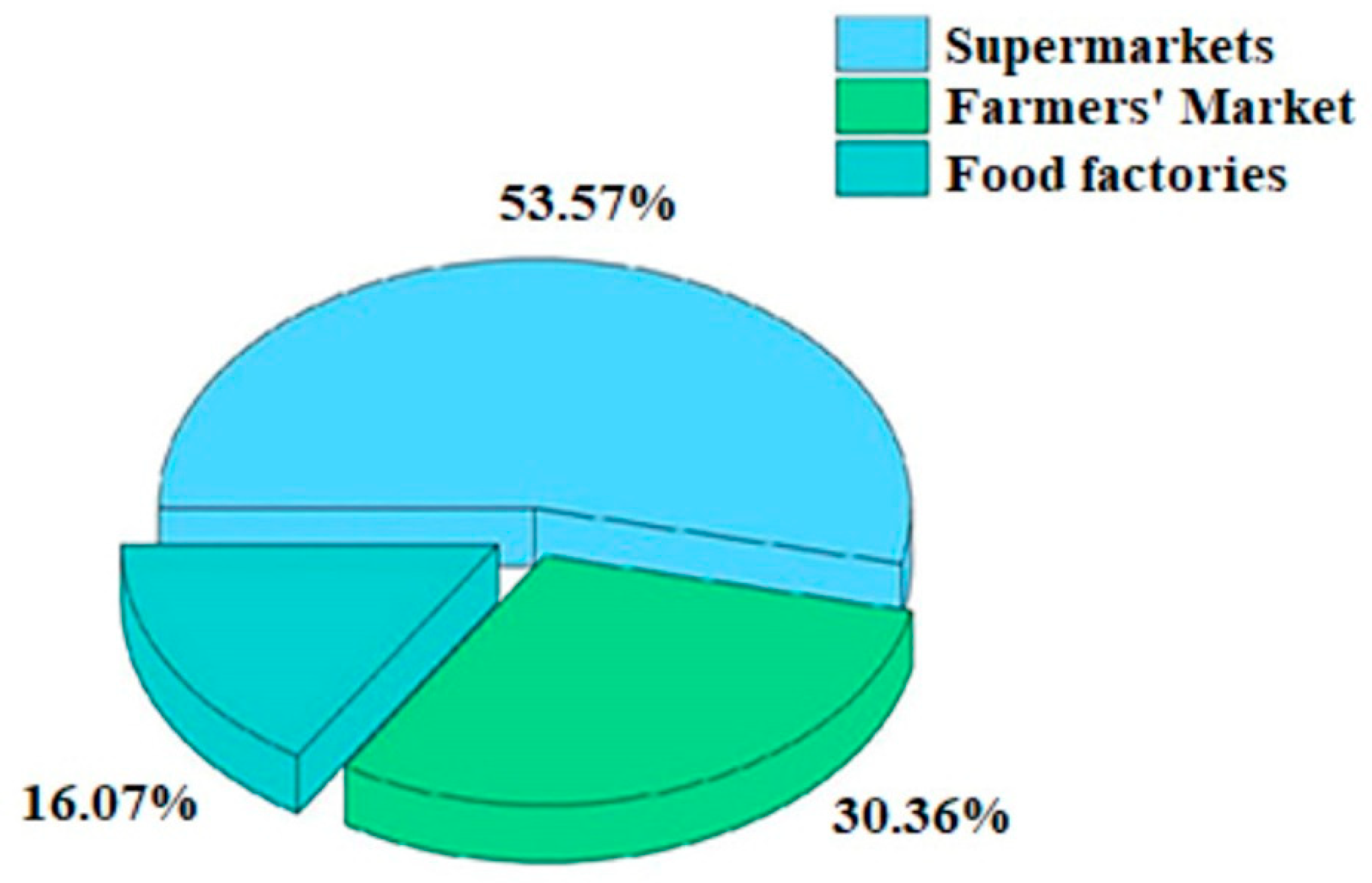
| Samples | Supermarkets | Farmers’ Markets | Food Factories | Total | Pollution Rate (%) |
|---|---|---|---|---|---|
| Wild mushrooms | 4 | 6 | — | 10/32 | 31.25 |
| Soybean products | 3 | 3 | 2 | 9/76 | 11.84 |
| Fresh vegetables | 10 | 2 | — | 12/80 | 15.00 |
| Pickled vegetables | 4 | 4 | 3 | 11/47 | 23.40 |
| Quick-freeze food | 9 | 2 | — | 10/34 | 29.41 |
| Cereals | — | — | 4 | 4/4 | 100.00 |
| Total | 30/180 | 17/81 | 9/12 | 56/273 | 20.51 |
| Samples | Supermarkets | Farmers’ Markets | Food Factories | Total | Pollution Rate (%) |
|---|---|---|---|---|---|
| Area A | 6 | — | — | 6/14 | 42.59 |
| Area B | 7 | 3 | — | 10/33 | 30.30 |
| Area C | — | 5 | — | 5/12 | 41.67 |
| Area D | 8 | 8 | — | 16/58 | 27.59 |
| Area E | 5 | — | — | 5/49 | 10.20 |
| Area F | 0 | 0 | — | 0/53 | 0.00 |
| Area G | 4 | 1 | — | 5/42 | 11.90 |
| Area H | — | — | 9 | 9/12 | 75.00 |
| Total | 30/180 | 17/81 | 9/12 | 56/273 | 20.51 |
| Type ST | glp | gmk | ilv | pta | pur | pyc | tpi | Number (Plants) | Clonal_Complex |
|---|---|---|---|---|---|---|---|---|---|
| 4 | 13 | 8 | 8 | 11 | 11 | 12 | 7 | 1 | ST-142 complex |
| 24 | 12 | 8 | 9 | 14 | 11 | 12 | 10 | 1 | — |
| 26 | 3 | 2 | 31 | 5 | 16 | 3 | 4 | 11 | — |
| 32 | 5 | 4 | 3 | 4 | 15 | 6 | 16 | 1 | — |
| 90 | 6 | 4 | 41 | 5 | 43 | 46 | 3 | 3 | — |
| 92 | 6 | 4 | 42 | 4 | 16 | 6 | 3 | 3 | — |
| 111 | 43 | 26 | 35 | 42 | 39 | 41 | 30 | 1 | ST-111 complex |
| 114 | 8 | 10 | 105 | 36 | 17 | 70 | 11 | 1 | — |
| 164 | 3 | 2 | 63 | 5 | 36 | 3 | 4 | 3 | — |
| 172 | 6 | 4 | 3 | 63 | 16 | 6 | 3 | 1 | — |
| 177 | 13 | 47 | 9 | 11 | 68 | 12 | 10 | 5 | — |
| 191 | 15 | 6 | 29 | 8 | 4 | 7 | 7 | 1 | ST-97 complex |
| 233 | 87 | 26 | 91 | 90 | 91 | 75 | 30 | 1 | — |
| 549 | 3 | 2 | 59 | 17 | 19 | 126 | 55 | 2 | — |
| 761 | 19 | 2 | 59 | 65 | 19 | 3 | 55 | 1 | ST-205 complex |
| 1000 | 13 | 8 | 8 | 58 | 122 | 12 | 7 | 1 | ST-142 complex |
| 1065 | 3 | 2 | 31 | 348 | 49 | 3 | 2 | 1 | ST-205 complex |
| 1120 | 19 | 2 | 31 | 17 | 19 | 3 | 2 | 1 | ST-205 complex |
| 1223 | 55 | 1 | 83 | 1 | 230 | 37 | 43 | 1 | — |
| 1259 | 14 | 8 | 9 | 11 | 9 | 88 | 8 | 3 | — |
| 1419 | 94 | 2 | 154 | 5 | 32 | 3 | 114 | 2 | — |
| 1431 | 13 | 8 | 8 | 11 | 9 | 12 | 10 | 8 | ST-142 complex |
| 1443 | 19 | 2 | 234 | 5 | 208 | 18 | 2 | 1 | ST-205 complex |
| 1599 | 19 | 2 | 234 | 5 | 19 | 18 | 2 | 1 | ST-205 complex |
| 1615 | 48 | 30 | 33 | 37 | 44 | 31 | 51 | 1 | — |
| 1667 | 53 | 2 | 59 | 5 | 47 | 3 | 216 | 4 | — |
| 1941 | 3 | 2 | 31 | 5 | 36 | 3 | 4 | 1 | — |
| 1989 | 324 | 2 | 122 | 5 | 19 | 3 | 91 | 1 | ST-205 complex |
| 2251 | 53 | 2 | 59 | 17 | 19 | 3 | 2 | 3 | ST-205 complex |
| 2431 | 13 | 47 | 9 | 11 | 68 | 3 | 10 | 3 | — |
| 2491 | 13 | 8 | 8 | 11 | 122 | 12 | 8 | 1 | ST-142 complex |
| 2587 | 239 | 142 | 269 | 180 | 237 | 149 | 37 | 2 | — |
| 2700 | 226 | 31 | 231 | 43 | 45 | 53 | 159 | 1 | — |
| 2718 | 61 | 38 | 1 | 1 | 18 | 37 | 19 | 1 | — |
| Genotypes | Toxic Genes | Number of Carrier Strains (Strains) | Detection Rates (%) |
|---|---|---|---|
| Vomitoxins | cesA | 16 | 21.92 |
| cesB | 15 | 20.55 | |
| cesC | 16 | 21.92 | |
| cesD | 16 | 21.92 | |
| cesH | 24 | 32.88 | |
| cesP | 16 | 21.92 | |
| cesT | 16 | 21.92 | |
| Hemolytic enterotoxins | HblA | 34 | 46.58 |
| HblC | 34 | 46.58 | |
| HblD | 34 | 46.58 | |
| Non-hemolytic enterotoxins | NheA | 72 | 98.63 |
| NheB | 73 | 100 | |
| NheC | 73 | 100 | |
| Cytotoxins | CytK | 46 | 63.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Cha, X.; Brennan, C.; Cao, J.; Shang, Y. Contamination of Plant Foods with Bacillus cereus in a Province and Analysis of Its Traceability. Microorganisms 2023, 11, 2763. https://doi.org/10.3390/microorganisms11112763
Lin Y, Cha X, Brennan C, Cao J, Shang Y. Contamination of Plant Foods with Bacillus cereus in a Province and Analysis of Its Traceability. Microorganisms. 2023; 11(11):2763. https://doi.org/10.3390/microorganisms11112763
Chicago/Turabian StyleLin, Yingting, Xiaoyan Cha, Charles Brennan, Jianxin Cao, and Ying Shang. 2023. "Contamination of Plant Foods with Bacillus cereus in a Province and Analysis of Its Traceability" Microorganisms 11, no. 11: 2763. https://doi.org/10.3390/microorganisms11112763
APA StyleLin, Y., Cha, X., Brennan, C., Cao, J., & Shang, Y. (2023). Contamination of Plant Foods with Bacillus cereus in a Province and Analysis of Its Traceability. Microorganisms, 11(11), 2763. https://doi.org/10.3390/microorganisms11112763