
Anomaly Detection Models for SARS-CoV-2 Surveillance Based on Genome k-mers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Data Processing
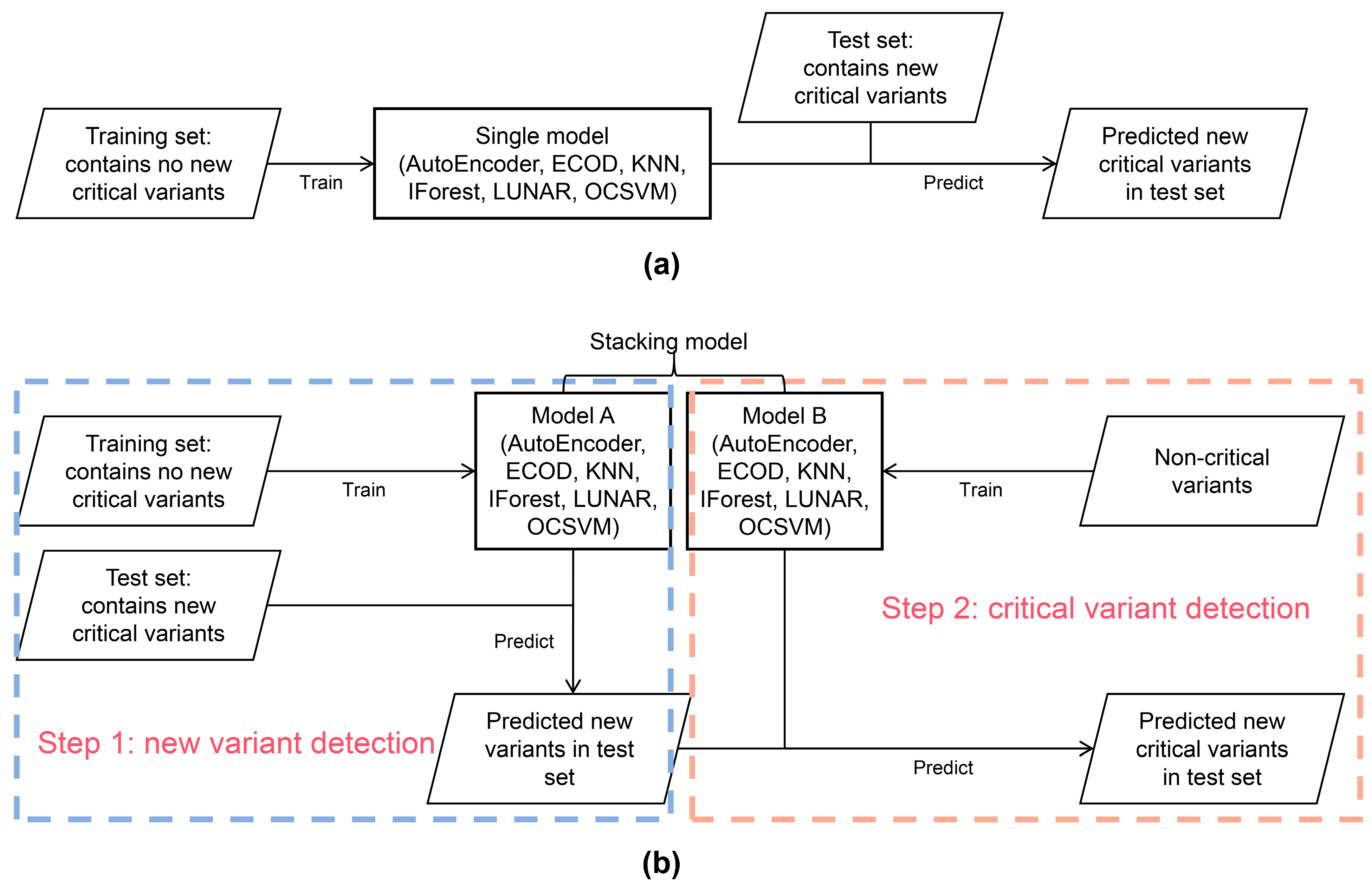
2.3. Anomaly Detection Models
2.4. Dataset Preparation
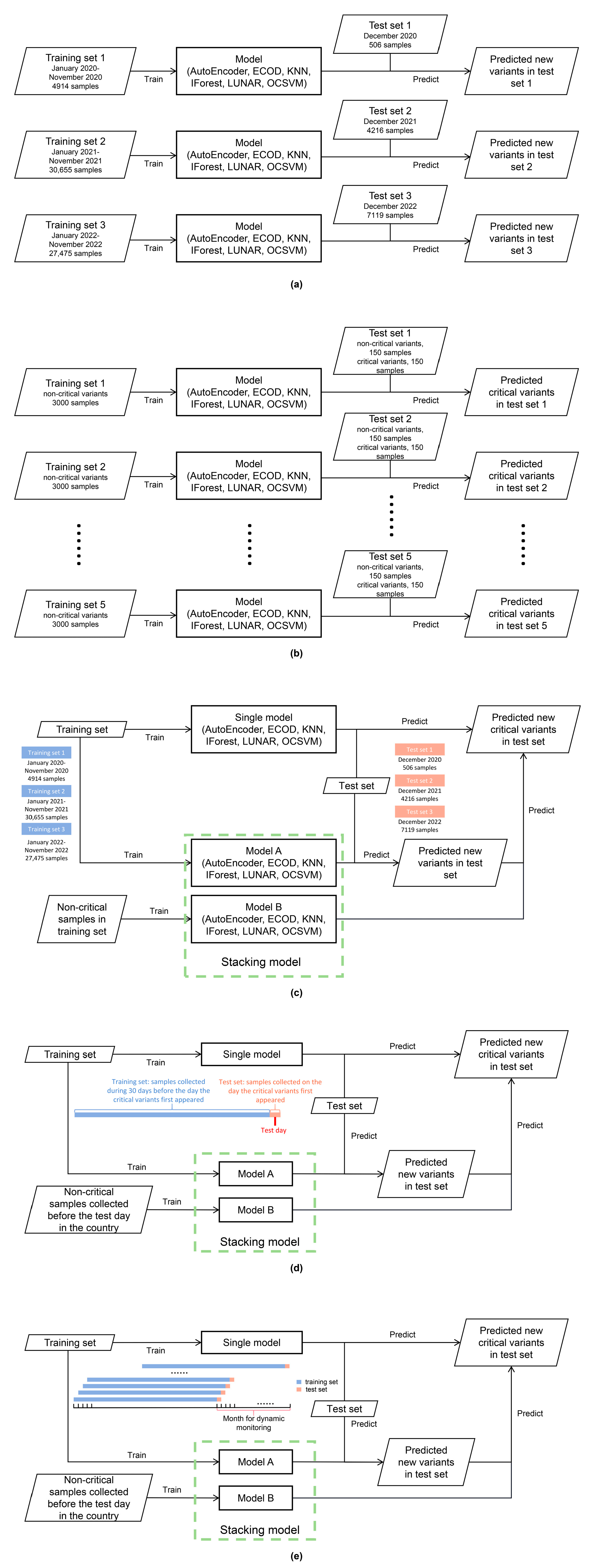
2.4.1. Datasets for Model Evaluation of New Variant Detection
2.4.2. Datasets for Model Evaluation of Critical Variant Detection
2.4.3. Datasets for Model Evaluation of New Critical Variant Detection
2.4.4. Datasets for Comparing the Detection of All Critical Variants on the Days They First Appeared in Three Countries
2.4.5. Datasets for Analog Dynamic Monitoring
3. Results
3.1. Evaluation of New Variant Detection
3.2. Evaluation of Critical Variant Detection
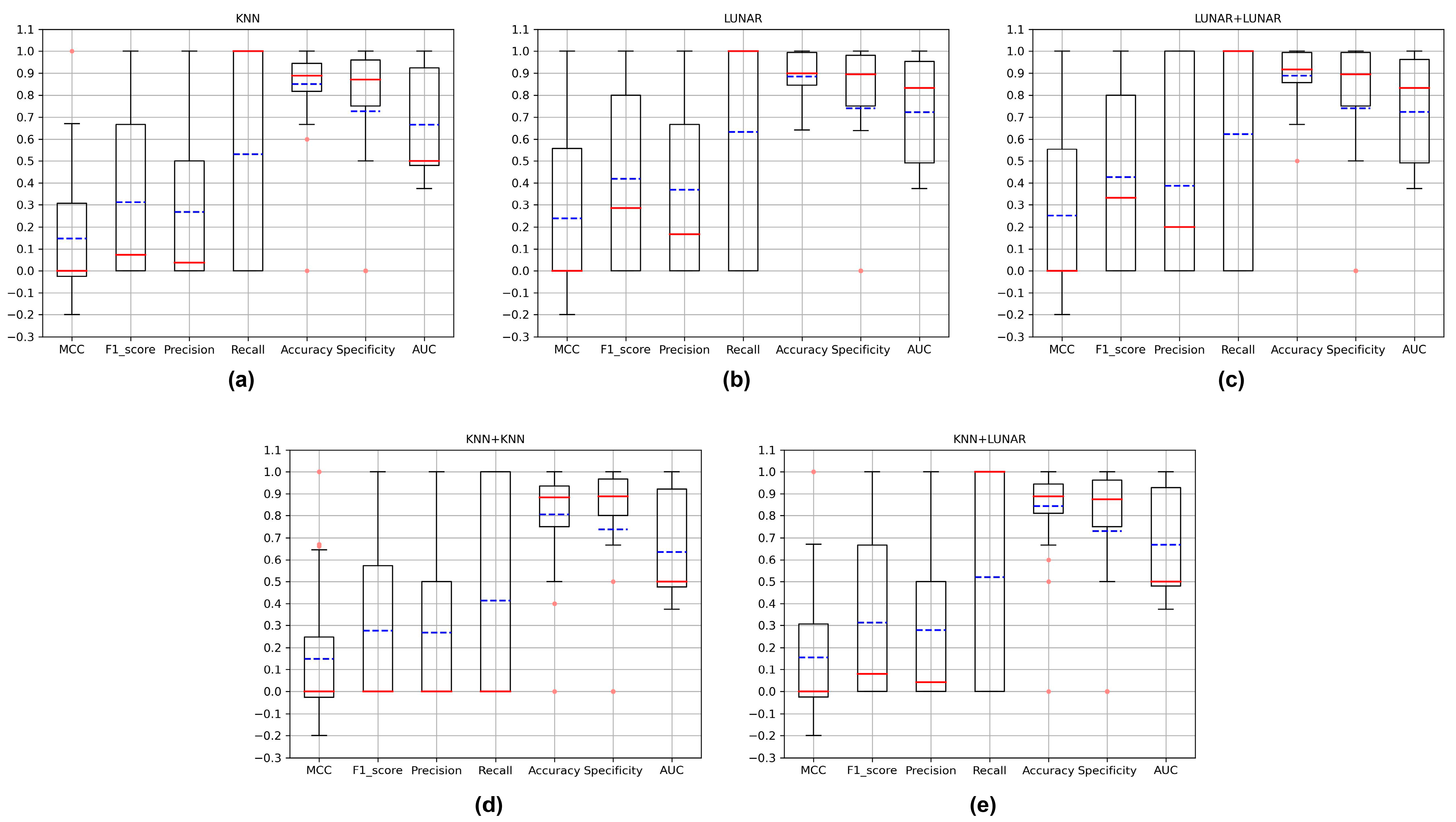
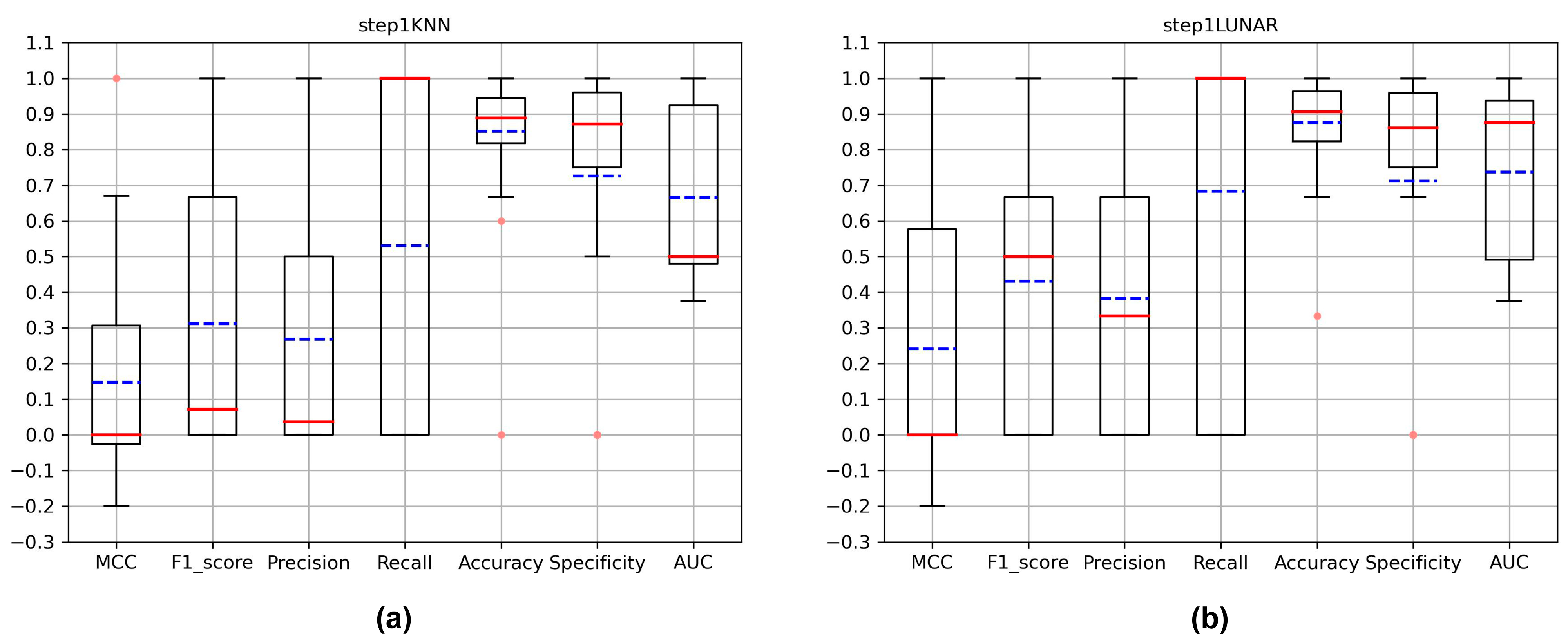
3.3. Evaluation of New Critical Variant Detection
3.4. Comparing the Detection of All Critical Variants on the Days They First Appeared in Three Countries
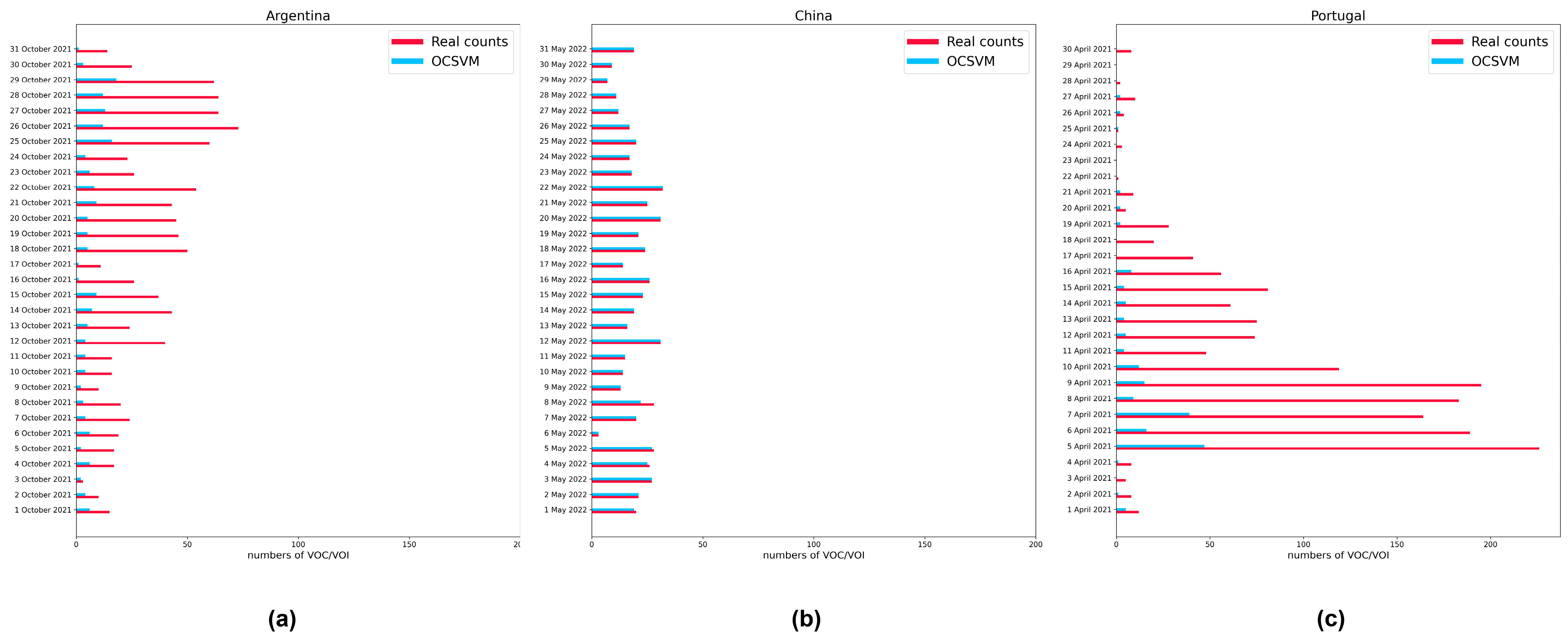
3.5. Analog Dynamic Monitoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, T.; Huang, T.; Guo, C.; Wang, A.; Shi, X.; Mo, X.; Lu, Q.; Sun, J.; Hui, T.; Tian, G.; et al. Genomic Variation, Origin Tracing, and Vaccine Development of SARS-CoV-2: A Systematic Review. Innovation 2021, 2, 100116. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 8 May 2023).
- Ren, S.Y.; Wang, W.B.; Gao, R.D.; Zhou, A.M. Omicron Variant (B.1.1.529) of SARS-CoV-2: Mutation, Infectivity, Transmission, and Vaccine Resistance. World J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Al-Thani, H.; El-Menyar, A. The Emergence of New SARS-CoV-2 Variant (Omicron) and Increasing Calls for COVID-19 vaccine boosters-The debate continues. Travel Med. Infect. Dis. 2022, 45, 102246. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chang, L.; Wang, L.N. Laboratory testing of SARS-CoV, MERS-CoV, and SARS-CoV-2 (2019-nCoV): Current Status, Challenges, and Countermeasures. Rev. Med. Virol. 2020, 30, e2106. [Google Scholar] [CrossRef] [PubMed]
- Goswami, C.; Sheldon, M.; Bixby, C.; Keddache, M.; Bogdanowicz, A.; Wang, Y.H.; Schultz, J.; McDevitt, J.; LaPorta, J.; Kwon, E.; et al. Identification of SARS-CoV-2 Variants Using Viral Sequencing for the Centers for Disease Control and Prevention Genomic Surveillance Program. BMC Infect. Dis. 2022, 22, 1–12. [Google Scholar] [CrossRef]
- Berno, G.; Fabeni, L.; Matusali, G.; Gruber, C.E.M.; Rueca, M.; Giombini, E.; Garbuglia, A.R. SARS-CoV-2 Variants Identification: Overview of Molecular Existing Methods. Pathogens 2022, 11, 1058. [Google Scholar] [CrossRef]
- Caputo, E.; Mandrich, L. Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life 2022, 12, 1245. [Google Scholar] [CrossRef]
- Ren, H.; Ling, Y.; Cao, R.; Wang, Z.; Li, Y.; Huang, T. Early Warning of Emerging Infectious Diseases Based on Multimodal Data. Biosaf. Health 2023. online ahead of print. [Google Scholar] [CrossRef]
- Chao, J.N.; Tang, F.R.; Xu, L. Developments in Algorithms for Sequence Alignment: A Review. Biomolecules 2022, 12, 546. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Chatzou, M.; Magis, C.; Chang, J.M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple Sequence Alignment Modeling: Methods and Applications. Brief. Bioinform. 2016, 17, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-Free Sequence Comparison: Benefits, Applications, and Tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) Enables Real-Time Phylogenetics for the SARS-CoV-2 Pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Forsdyke, D.R. Success of Alignment-Free Oligonucleotide (k-mer) Analysis Confirms Relative Importance of Genomes not Genes in Speciation and Phylogeny. Biol. J. Linn. Soc. 2019, 128, 239–250. [Google Scholar] [CrossRef]
- Li, W.T.; Freudenberg, J.; Freudenberg, J. Alignment-Free Approaches for Predicting Novel Nuclear Mitochondrial Segments (NUMTs) in the Human Genome. Gene 2019, 691, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.L.; Yu, Z.G.; Tang, R.B.; Xie, X.H.; Han, G.H.; Anh, V.V. Phylogenetic Analysis of HIV-1 Genomes Based on the Position-Weighted K-mers Method. Entropy 2020, 22, 255. [Google Scholar] [CrossRef]
- Petrucci, E.; Noé, L.; Pizzi, C.; Comin, M. Iterative Spaced Seed Hashing: Closing the Gap Between Spaced Seed Hashing and k-mer Hashing. Bioinform. Res. Appl. Isbra 2019, 11490, 208–219. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F.Z. VirFinder: A Novel k-mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 1–20. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Tang, K.J.; Ren, J.; Fuhrman, J.A.; Waterman, M.S.; Sun, F.Z. CAFE: Accelerated Alignment-FrEe Sequence Analysis. Nucleic Acids Res. 2017, 45, W554–W559. [Google Scholar] [CrossRef]
- Ren, R.H.; Yin, C.C.; Yau, S.S.T. kmer2vec: A Novel Method for Comparing DNA Sequences by word2vec Embedding. J. Comput. Biol. 2022, 29, 1001–1021. [Google Scholar] [CrossRef]
- Tang, R.B.; Yu, Z.G.; Li, J.Y. KINN: An Alignment-Free Accurate Phylogeny Reconstruction Method Based on Inner Distance Distributions of k-mer Pairs in Biological Sequences. Mol. Phylogenet Evol. 2023, 179, 107662. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zhang, Y.; Yau, S.S. k-mer Sparse Matrix Model for Genetic Sequence and Its Applications in Sequence Comparison. J. Theor. Biol. 2014, 363, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Nassif, A.B.; Talib, M.A.; Nasir, Q.; Dakalbab, F.M. Machine Learning for Anomaly Detection: A Systematic Review. IEEE Access 2021, 9, 78658–78700. [Google Scholar] [CrossRef]
- Zimek, A.; Filzmoser, P. There and Back Again: Outlier Detection between Statistical Reasoning and Data Mining Algorithms. Wires Data Min. Knowl. 2018, 8, e1280. [Google Scholar] [CrossRef]
- Pimentel, M.A.F.; Clifton, D.A.; Clifton, L.; Tarassenko, L. A Review of Novelty Detection. Signal Process. 2014, 99, 215–249. [Google Scholar] [CrossRef]
- Chandola, V.; Banerjee, A.; Kumar, V. Anomaly Detection: A survey. ACM Comput. Surv. 2009, 41, 1–58. [Google Scholar] [CrossRef]
- Pang, G.S.; Shen, C.H.; Cao, L.B.; Van den Hengel, A. Deep Learning for Anomaly Detection: A Review. ACM Comput. Surv. 2021, 54, 1–38. [Google Scholar] [CrossRef]
- Zhao, Y.; Nasrullah, Z.; Li, Z. PyOD: A Python Toolbox for Scalable Outlier Detection. Comput. Sci. 2019, 20, 1–7. [Google Scholar]
- Li, Z.; Zhao, Y.; Hu, X.; Botta, N.; Ionescu, C.; Chen, G. ECOD: Unsupervised Outlier Detection Using Empirical Cumulative Distribution Functions. IEEE Trans. Knowl. Data Eng. 2022, 35, 12181–12193. [Google Scholar] [CrossRef]
- Schölkopf, B.; Williamson, R.; Smola, A.; Shawe-Taylor, J.; Platt, J. Support Vector Method for Novelty Detection. In Proceedings of the 12th International Conference on Neural Information Processing Systems, Denver, CO, USA, 29 November–4 December 1999; pp. 582–588. [Google Scholar]
- Angiulli, F.; Pizzuti, C. Fast Outlier Detection in High Dimensional Spaces. In Principles of Data Mining and Knowledge Discovery; Springer: Berlin/Heidelberg, Germany, 2002; pp. 15–27. [Google Scholar]
- Liu, F.T.; Ting, K.M.; Zhou, Z.-H. Isolation-Based Anomaly Detection. ACM Trans. Knowl. Discov. Data 2012, 6, 1–39. [Google Scholar] [CrossRef]
- Michelucci, U. Autoencoders. In Applied Deep Learning with TensorFlow 2: Learn to Implement Advanced Deep Learning Techniques with Python; Apress: Berkeley, CA, USA, 2022; pp. 257–283. [Google Scholar] [CrossRef]
- Goodge, A.; Hooi, B.; Ng, S.K.; Ng, W.S. LUNAR: Unifying Local Outlier Detection Methods via Graph Neural Networks. AAAI Conf. Artif. Intell. 2022, 36, 6737–6745. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Liu, J.J.; Shen, W.J. A Review of Ensemble Learning Algorithms Used in Remote Sensing Applications. Appl. Sci. 2022, 12, 8654. [Google Scholar] [CrossRef]
- van Engelen, J.E.; Hoos, H.H. A Survey on Semi-Supervised Learning. Mach. Learn. 2020, 109, 373–440. [Google Scholar] [CrossRef]
- Nicora, G.; Salemi, M.; Marini, S.; Bellazzi, R. Predicting Emerging SARS-CoV-2 Variants of Concern through a One Class Dynamic Anomaly Detection Algorithm. BMJ Health Care Inform. 2022, 29, e100643. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Taskforce for Joint Prevention and Control Mechanism for COVID-19 under the State Council and the National Health Commission of P.R. China. Scientific and Targeted Prevention and Control Measures to Optimize COVID-19 Response. Health Care Sci. 2023, 2, 1–6. [Google Scholar] [CrossRef]
- Milhinhos, A.; Costa, P.M. On the Progression of COVID-19 in Portugal: A Comparative Analysis of Active Cases Using Non-linear Regression. Front. Public Health 2020, 8, 495. [Google Scholar] [CrossRef]
- Larrosa, J.M.C. SARS-CoV-2 in Argentina: Lockdown, Mobility, and Contagion. J. Med. Virol. 2021, 93, 2252–2261. [Google Scholar] [CrossRef]
- Data, Our World in “Coronavirus (COVID-19)” Vaccinations. Available online: https://ourworldindata.org/covid-vaccinations (accessed on 21 October 2023).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The Advantages of the Matthews Correlation Coefficient (MCC) over F1 Score and Accuracy in Binary Classification Evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef]
- Markus, M.T.; Groenen, P.J.F. An Introduction to the Bootstrap. Psychometrika 1998, 63, 97–101. [Google Scholar]
- Harrison, A.G.; Lin, T.; Wang, P.H. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L.; Budd, A.; Cox, N. Seasonal and Pandemic Influenza Surveillance Considerations for Constructing Multicomponent Systems. Influenza Other Respir. Viruses 2009, 3, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bonidia, R.P.; Domingues, D.S.; Sanches, D.S.; de Carvalho, A. MathFeature: Feature Extraction Package for DNA, RNA and Protein Sequences Based on Mathematical Descriptors. Brief. Bioinform. 2022, 23, bbab434. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Lee, V.C.; Salehi, M. MIR_MAD: An Efficient and On-line Approach for Anomaly Detection in Dynamic Data Stream. In Proceedings of the 20th IEEE International Conference on Data Mining (ICDM), Electr Network, Sorrento, Italy, 17–20 November 2020; pp. 424–431. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | MCC | F1-Score | Precision | Recall | Accuracy | Specificity | AUC |
|---|---|---|---|---|---|---|---|
| AutoEncoder [34] | −0.016 | 0.004 | 0.002 | 0.078 | 0.838 | 0.842 | 0.460 |
| ECOD [30] | −0.005 | 0.008 | 0.004 | 0.078 | 0.887 | 0.891 | 0.485 |
| IForest [33] | −0.012 | 0.005 | 0.003 | 0.078 | 0.857 | 0.860 | 0.469 |
| KNN [32] | 0.092 | 0.035 | 0.018 | 0.741 | 0.793 | 0.792 | 0.767 |
| LUNAR [35] | 0.080 | 0.036 | 0.019 | 0.556 | 0.837 | 0.838 | 0.697 |
| OCSVM [31] | 0.006 | 0.010 | 0.005 | 0.207 | 0.874 | 0.878 | 0.543 |
| Model | MCC | F1-Score | Precision | Recall | Accuracy | Specificity | AUC |
|---|---|---|---|---|---|---|---|
| AutoEncoder [34] | 0.539 | 0.722 | 0.855 | 0.625 | 0.759 | 0.893 | 0.759 |
| ECOD [30] | 0.122 | 0.303 | 0.643 | 0.200 | 0.544 | 0.889 | 0.544 |
| IForest [33] | 0.192 | 0.372 | 0.697 | 0.257 | 0.576 | 0.895 | 0.576 |
| KNN [32] | 0.171 | 0.345 | 0.691 | 0.231 | 0.564 | 0.897 | 0.564 |
| LUNAR [35] | 0.663 | 0.811 | 0.891 | 0.745 | 0.827 | 0.908 | 0.827 |
| OCSVM [31] | −0.009 | 0.174 | 0.487 | 0.107 | 0.497 | 0.888 | 0.497 |
| Model | MCC | F1-Score | Precision | Recall | Accuracy | Specificity | AUC |
|---|---|---|---|---|---|---|---|
| AutoEncoder [34] | −0.014 | 0.004 | 0.002 | 0.078 | 0.839 | 0.842 | 0.460 |
| ECOD [30] | −0.003 | 0.008 | 0.004 | 0.078 | 0.888 | 0.891 | 0.485 |
| IForest [33] | −0.008 | 0.006 | 0.003 | 0.098 | 0.850 | 0.853 | 0.476 |
| KNN [32] | 0.105 | 0.035 | 0.018 | 0.852 | 0.794 | 0.793 | 0.822 |
| LUNAR [35] | 0.104 | 0.046 | 0.024 | 0.722 | 0.811 | 0.811 | 0.767 |
| OCSVM [31] | 0.008 | 0.010 | 0.005 | 0.207 | 0.875 | 0.878 | 0.543 |
| AutoEncoder+AutoEncoder | −0.011 | 0.004 | 0.002 | 0.078 | 0.860 | 0.862 | 0.470 |
| AutoEncoder+ECOD | −0.016 | 0.001 | 0.001 | 0.010 | 0.920 | 0.924 | 0.467 |
| AutoEncoder+IForest | −0.005 | 0.006 | 0.003 | 0.078 | 0.890 | 0.893 | 0.486 |
| AutoEncoder+KNN | −0.010 | 0.004 | 0.002 | 0.078 | 0.864 | 0.867 | 0.473 |
| AutoEncoder+LUNAR | −0.011 | 0.004 | 0.002 | 0.078 | 0.861 | 0.864 | 0.471 |
| AutoEncoder+OCSVM | −0.006 | 0.007 | 0.004 | 0.069 | 0.887 | 0.891 | 0.480 |
| ECOD+AutoEncoder | 0.000 | 0.008 | 0.004 | 0.078 | 0.907 | 0.910 | 0.494 |
| ECOD+ECOD | −0.013 | 0.003 | 0.002 | 0.020 | 0.919 | 0.923 | 0.471 |
| ECOD+IForest | 0.000 | 0.008 | 0.004 | 0.078 | 0.906 | 0.909 | 0.494 |
| ECOD+KNN | 0.000 | 0.008 | 0.004 | 0.078 | 0.908 | 0.911 | 0.495 |
| ECOD+LUNAR | 0.000 | 0.008 | 0.004 | 0.078 | 0.907 | 0.910 | 0.494 |
| ECOD+OCSVM | −0.005 | 0.007 | 0.004 | 0.069 | 0.894 | 0.897 | 0.483 |
| IForest+AutoEncoder | −0.002 | 0.007 | 0.004 | 0.078 | 0.902 | 0.905 | 0.492 |
| IForest+ECOD | −0.015 | 0.001 | 0.001 | 0.010 | 0.921 | 0.925 | 0.467 |
| IForest+IForest | −0.003 | 0.007 | 0.004 | 0.069 | 0.905 | 0.909 | 0.489 |
| IForest+KNN | −0.004 | 0.007 | 0.003 | 0.098 | 0.878 | 0.881 | 0.489 |
| IForest+LUNAR | 0.000 | 0.008 | 0.004 | 0.088 | 0.900 | 0.903 | 0.496 |
| IForest+OCSVM | −0.006 | 0.007 | 0.004 | 0.069 | 0.888 | 0.891 | 0.480 |
| KNN+AutoEncoder | −0.002 | 0.007 | 0.004 | 0.108 | 0.876 | 0.879 | 0.493 |
| KNN+ECOD | −0.016 | 0.001 | 0.001 | 0.010 | 0.920 | 0.924 | 0.467 |
| KNN+IForest | −0.006 | 0.006 | 0.003 | 0.078 | 0.888 | 0.891 | 0.485 |
| KNN+KNN | 0.081 | 0.031 | 0.016 | 0.578 | 0.864 | 0.864 | 0.721 |
| KNN+LUNAR | 0.064 | 0.025 | 0.013 | 0.500 | 0.845 | 0.845 | 0.672 |
| KNN+OCSVM | 0.011 | 0.010 | 0.005 | 0.254 | 0.861 | 0.864 | 0.559 |
| LUNAR+AutoEncoder | 0.008 | 0.012 | 0.006 | 0.108 | 0.907 | 0.910 | 0.509 |
| LUNAR+ECOD | −0.010 | 0.005 | 0.002 | 0.029 | 0.918 | 0.921 | 0.475 |
| LUNAR+IForest | −0.004 | 0.006 | 0.003 | 0.069 | 0.901 | 0.904 | 0.486 |
| LUNAR+KNN | 0.075 | 0.037 | 0.020 | 0.412 | 0.909 | 0.910 | 0.661 |
| LUNAR+LUNAR | 0.110 | 0.047 | 0.025 | 0.637 | 0.891 | 0.891 | 0.764 |
| LUNAR+OCSVM | 0.012 | 0.011 | 0.006 | 0.291 | 0.834 | 0.837 | 0.564 |
| OCSVM+AutoEncoder | −0.005 | 0.006 | 0.003 | 0.059 | 0.909 | 0.913 | 0.486 |
| OCSVM+ECOD | −0.013 | 0.003 | 0.002 | 0.020 | 0.921 | 0.925 | 0.472 |
| OCSVM+IForest | −0.005 | 0.006 | 0.003 | 0.059 | 0.910 | 0.913 | 0.486 |
| OCSVM+KNN | −0.005 | 0.006 | 0.003 | 0.059 | 0.911 | 0.914 | 0.486 |
| OCSVM+LUNAR | −0.005 | 0.006 | 0.003 | 0.059 | 0.910 | 0.913 | 0.486 |
| OCSVM+OCSVM | 0.008 | 0.010 | 0.005 | 0.207 | 0.875 | 0.878 | 0.543 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, H.; Li, Y.; Huang, T. Anomaly Detection Models for SARS-CoV-2 Surveillance Based on Genome k-mers. Microorganisms 2023, 11, 2773. https://doi.org/10.3390/microorganisms11112773
Ren H, Li Y, Huang T. Anomaly Detection Models for SARS-CoV-2 Surveillance Based on Genome k-mers. Microorganisms. 2023; 11(11):2773. https://doi.org/10.3390/microorganisms11112773
Chicago/Turabian StyleRen, Haotian, Yixue Li, and Tao Huang. 2023. "Anomaly Detection Models for SARS-CoV-2 Surveillance Based on Genome k-mers" Microorganisms 11, no. 11: 2773. https://doi.org/10.3390/microorganisms11112773
APA StyleRen, H., Li, Y., & Huang, T. (2023). Anomaly Detection Models for SARS-CoV-2 Surveillance Based on Genome k-mers. Microorganisms, 11(11), 2773. https://doi.org/10.3390/microorganisms11112773