

Genomics of Yoonia sp. Isolates (Family Roseobacteraceae) from Lake Zhangnai on the Tibetan Plateau

Abstract
:1. Introduction
2. Methods
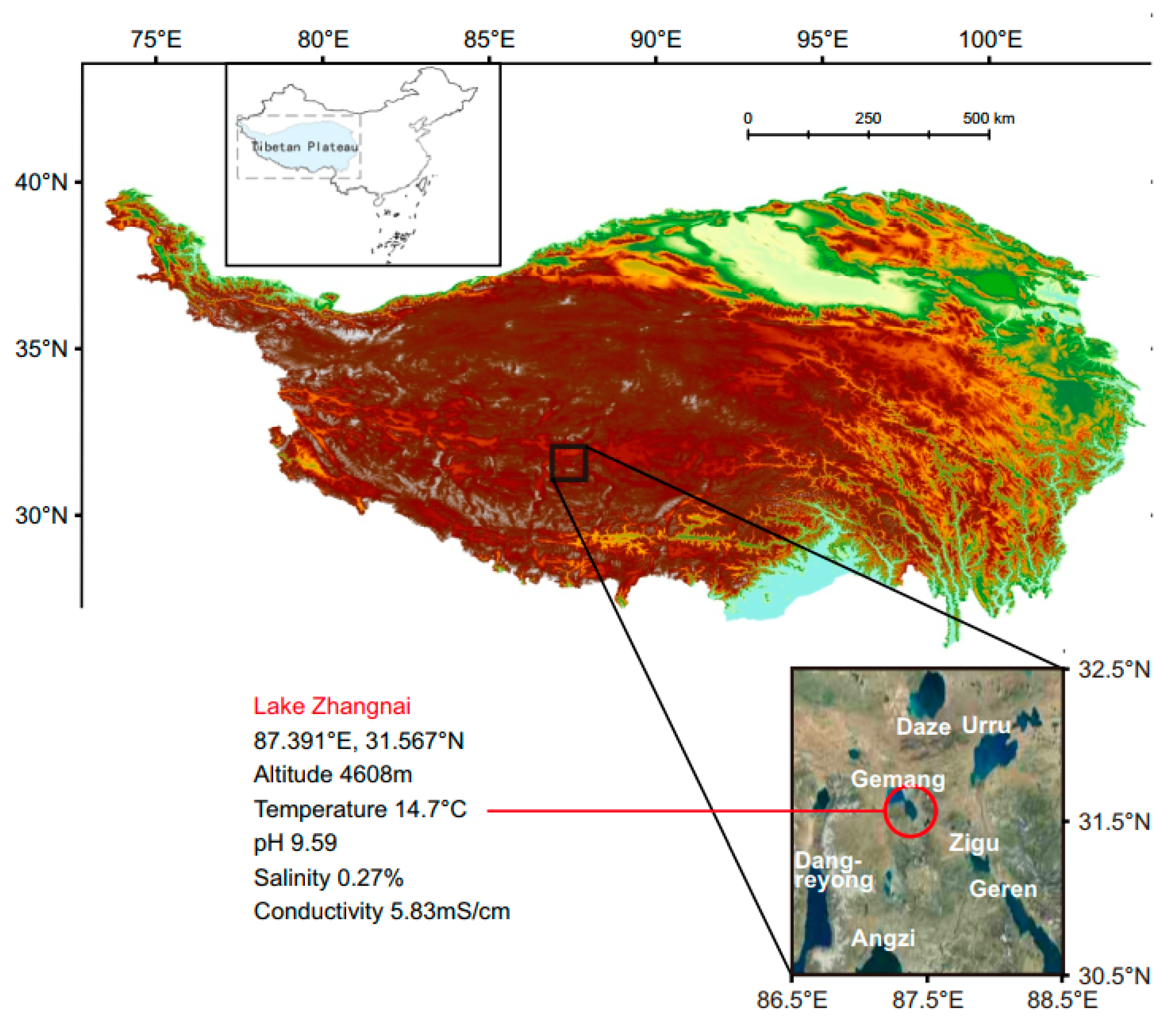
2.1. Sample Collection, Bacterial Cultivation, and Sequencing
2.2. Genomic Assembly and Annotation
2.3. Construction of Phylogenomic Trees
2.4. Metabolic Comparisons
3. Results and Discussion
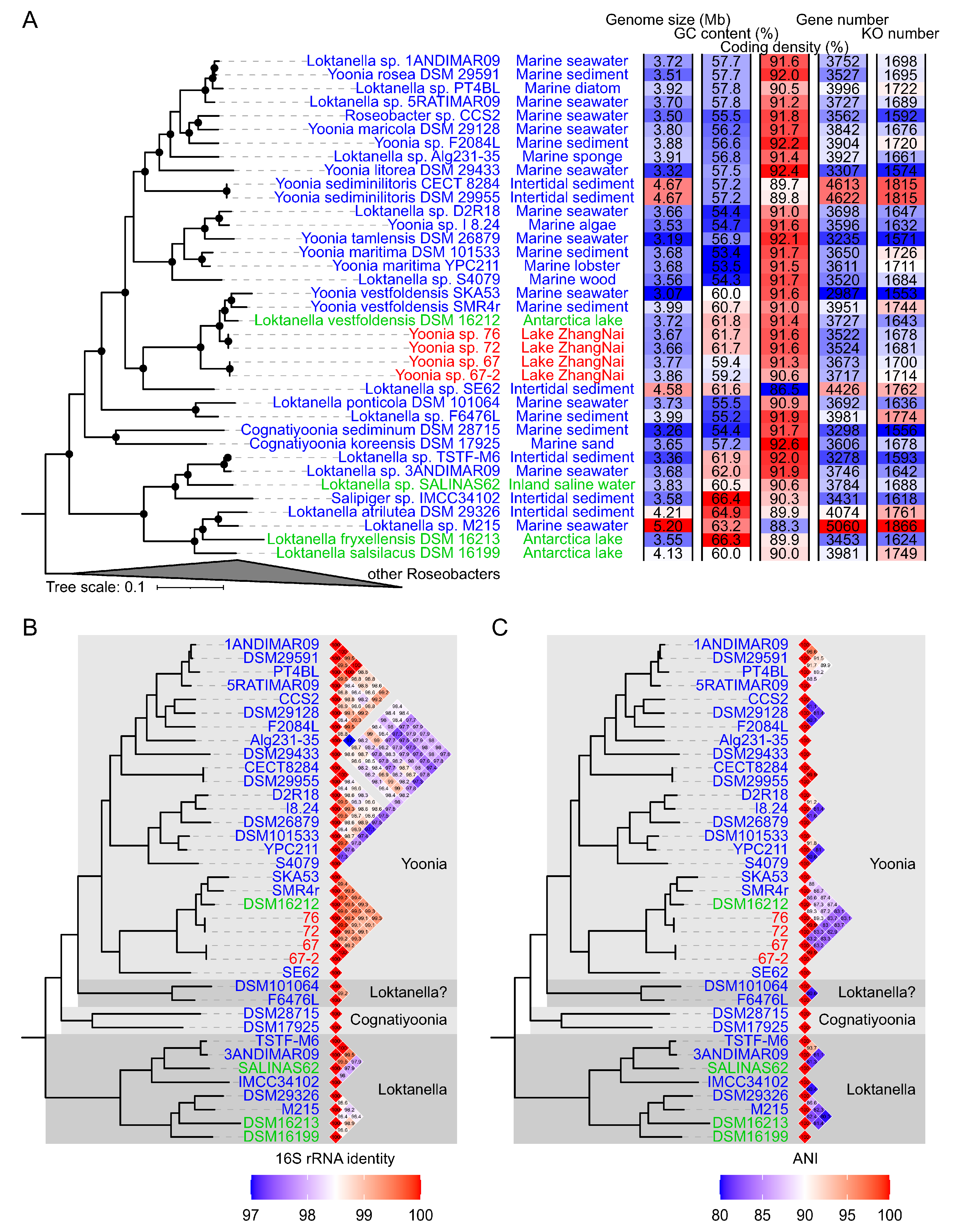
3.1. Genomic Characterization of Novel Roseobacteraceae Strains
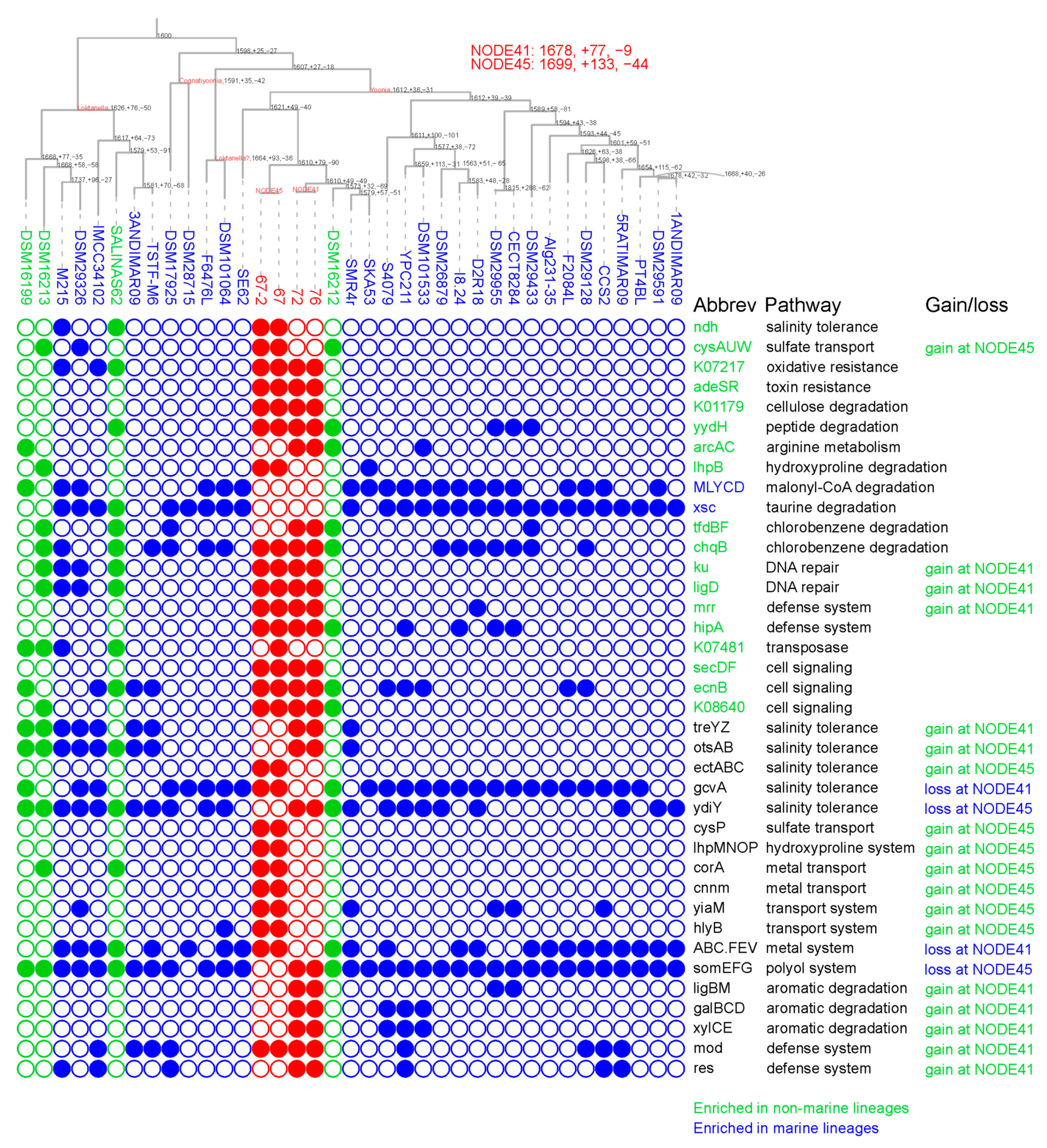
3.2. Habitat Transition from Marine to Lacustrine Environments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Logares, R.; Bråte, J.; Bertilsson, S.; Clasen, J.L.; Shalchian-Tabrizi, K.; Rengefors, K. Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol. 2009, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Paver, S.F.; Muratore, D.; Newton, R.J.; Coleman, M.L. Reevaluating the Salty Divide: Phylogenetic Specificity of Transitions between Marine and Freshwater Systems. mSystems 2018, 3, e00232-18. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Wang, J. Phylogenetic divergence and adaptation of Nitrososphaeria across lake depths and freshwater ecosystems. ISME J. 2022, 16, 1491–1501. [Google Scholar] [CrossRef]
- Henson, M.W.; Lanclos, V.C.; Faircloth, B.C.; Thrash, J.C. Cultivation and genomics of the first freshwater SAR11 (LD12) isolate. ISME J. 2018, 12, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; McLatchie, S.; Walsh, D.A. A Novel Freshwater to Marine Evolutionary Transition Revealed within Methylophilaceae Bacteria from the Arctic Ocean. mBio 2021, 12, e0130621. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Teng, W.-K.; Zhao, L.; Hu, C.-X.; Zhou, Y.-K.; Han, B.-P.; Song, L.-R.; Shu, W.-S. Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J. 2020, 15, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yoshizawa, S.; Sun, Y.; Huang, Y.; Chu, X.; González, J.M.; Pinhassi, J.; Luo, H. Repeated evolutionary transitions of flavobacteria from marine to non-marine habitats. Environ. Microbiol. 2019, 21, 648–666. [Google Scholar] [CrossRef]
- Cabello-Yeves, P.J.; Rodriguez-Valera, F. Marine-freshwater prokaryotic transitions require extensive changes in the predicted proteome. Microbiome 2019, 7, 117. [Google Scholar] [CrossRef]
- Chiriac, M.-C.; Haber, M.; Salcher, M.M. Adaptive genetic traits in pelagic freshwater microbes. Environ. Microbiol. 2023, 25, 606–641. [Google Scholar] [CrossRef]
- Moran, M.A.; Belas, R.; Schell, M.A.; González, J.M.; Sun, F.; Sun, S.; Binder, B.J.; Edmonds, J.; Ye, W.; Orcutt, B.; et al. Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol. 2007, 73, 4559–4569. [Google Scholar] [CrossRef]
- Liang, K.Y.H.; Orata, F.D.; Boucher, Y.F.; Case, R.J. Roseobacters in a Sea of Poly- and Paraphyly: Whole Genome-Based Taxonomy of the Family Rhodobacteraceae and the Proposal for the Split of the “Roseobacter Clade” into a Novel Family, Roseobacteraceae fam. nov. Front. Microbiol. 2021, 12, 683109. [Google Scholar] [CrossRef]
- Gifford, S.M.; Sharma, S.; Moran, M.A. Linking activity and function to ecosystem dynamics in a coastal bacterioplankton community. Front. Microbiol. 2014, 5, 185. [Google Scholar] [CrossRef]
- Wirth, J.S.; Whitman, W.B. Phylogenomic analyses of a clade within the Roseobacter group suggest taxonomic reassignments of species of the genera Aestuariivita, Citreicella, Loktanella, Nautella, Pelagibaca, Ruegeria, Thalassobius, Thiobacimonas and Tropicibacter, and the proposal of six novel genera. Int. J. Syst. Evol. Microbiol. 2018, 68, 2393–2411. [Google Scholar] [PubMed]
- Simon, M.; Scheuner, C.; Meier-Kolthoff, J.P.; Brinkhoff, T.; Wagner-Döbler, I.; Ulbrich, M.; Klenk, H.-P.; Schomburg, D.; Petersen, J.; Göker, M. Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 2017, 11, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Phurbu, D.; Wang, H.; Tang, Q.; Lu, H.; Zhu, H.; Jiang, S.; Xing, P.; Wu, Q.L. Tabrizicola alkalilacus sp. nov., isolated from alkaline Lake Dajiaco on the Tibetan Plateau. Int. J. Syst. Evol. Microbiol. 2019, 69, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Librado, P.; Vieira, F.G.; Rozas, J. BadiRate: Estimating family turnover rates by likelihood-based methods. Bioinformatics 2012, 28, 279–281. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Luo, H.; Moran, M.A. Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 2014, 78, 573–587. [Google Scholar] [CrossRef]
- van Trappen, S.; Mergaert, J.; Swings, J. Loktanella salsilacus gen. nov., sp. nov., Loktanella fryxellensis sp. nov. and Loktanella vestfoldensis sp. nov., new members of the Rhodobacter group, isolated from microbial mats in Antarctic lakes. Int. J. Syst. Evol. Microbiol. 2004, 54, 1263–1269. [Google Scholar] [CrossRef]
- Laniewski, K.; BorÉn, H.; Grimvall, A. Identification of Volatile and Extractable Chloroorganics in Rain and Snow. Environ. Sci. Technol. 1998, 32, 3935–3940. [Google Scholar] [CrossRef]
- Bremer, E.; Krämer, R. Responses of Microorganisms to Osmotic Stress. Annu. Rev. Microbiol. 2019, 73, 313–334. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Xiao, P.; Feng, X.; Li, H.; Ma, R.; Duan, H.; Zhao, L. Monitoring lake changes of Qinghai-Tibetan Plateau over the past 30 years using satellite remote sensing data. Chin. Sci. Bull. 2014, 59, 1021–1035. [Google Scholar] [CrossRef]
- Wetzel, R.G. Limnology: Lake and River Ecosystems; Gulf Professional Publishing: Huston, TX, USA, 2001. [Google Scholar]
- Sutak, R.; Camadro, J.-M.; Lesuisse, E. Iron Uptake Mechanisms in Marine Phytoplankton. Front. Microbiol. 2020, 11, 566691. [Google Scholar] [CrossRef]
- Jiang, H.; Lv, Q.; Yang, J.; Wang, B.; Dong, H.; Gonsior, M.; Schmitt-Kopplin, P. Molecular composition of dissolved organic matter in saline lakes of the Qing-Tibetan Plateau. Org. Geochem. 2022, 167, 104400. [Google Scholar] [CrossRef]
- Wang, J.; Fang, X.; Appel, E.; Song, C. Pliocene-Pleistocene Climate Change At the NE Tibetan Plateau Deduced From Lithofacies Variation In the Drill Core SG-1, Western Qaidam Basin, China. J. Sediment. Res. 2012, 82, 933–952. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genome ID | Contigs | N50 (bp) | Genome Size (bp) | GC Content | Coding Density | Genes |
|---|---|---|---|---|---|---|
| Yoonia sp. 72 | 10 | 770,970 | 3,664,109 | 61.7% | 91.6% | 3524 |
| Yoonia sp. 76 | 16 | 704,701 | 3,665,672 | 61.7% | 91.6% | 3522 |
| Yoonia sp. 67-2 | 80 | 299,240 | 3,855,349 | 59.2% | 90.6% | 3717 |
| Yoonia sp. 67 | 41 | 231,947 | 3,767,971 | 59.4% | 91.3% | 3673 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Xing, P. Genomics of Yoonia sp. Isolates (Family Roseobacteraceae) from Lake Zhangnai on the Tibetan Plateau. Microorganisms 2023, 11, 2817. https://doi.org/10.3390/microorganisms11112817
Feng X, Xing P. Genomics of Yoonia sp. Isolates (Family Roseobacteraceae) from Lake Zhangnai on the Tibetan Plateau. Microorganisms. 2023; 11(11):2817. https://doi.org/10.3390/microorganisms11112817
Chicago/Turabian StyleFeng, Xiaoyuan, and Peng Xing. 2023. "Genomics of Yoonia sp. Isolates (Family Roseobacteraceae) from Lake Zhangnai on the Tibetan Plateau" Microorganisms 11, no. 11: 2817. https://doi.org/10.3390/microorganisms11112817
APA StyleFeng, X., & Xing, P. (2023). Genomics of Yoonia sp. Isolates (Family Roseobacteraceae) from Lake Zhangnai on the Tibetan Plateau. Microorganisms, 11(11), 2817. https://doi.org/10.3390/microorganisms11112817