
MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods


Abstract
:1. Introduction
2. Materials and Methods
2.1. Random Forest vs. Gradient Boosting
2.2. Training Processes
2.3. R Libraries
2.4. Web Server and GitHub Repository
2.5. Data Availability
2.6. Code Availability
3. Results
3.1. Data Processing: Data Input, Quality Control and Data Transformation
3.2. Data Mining
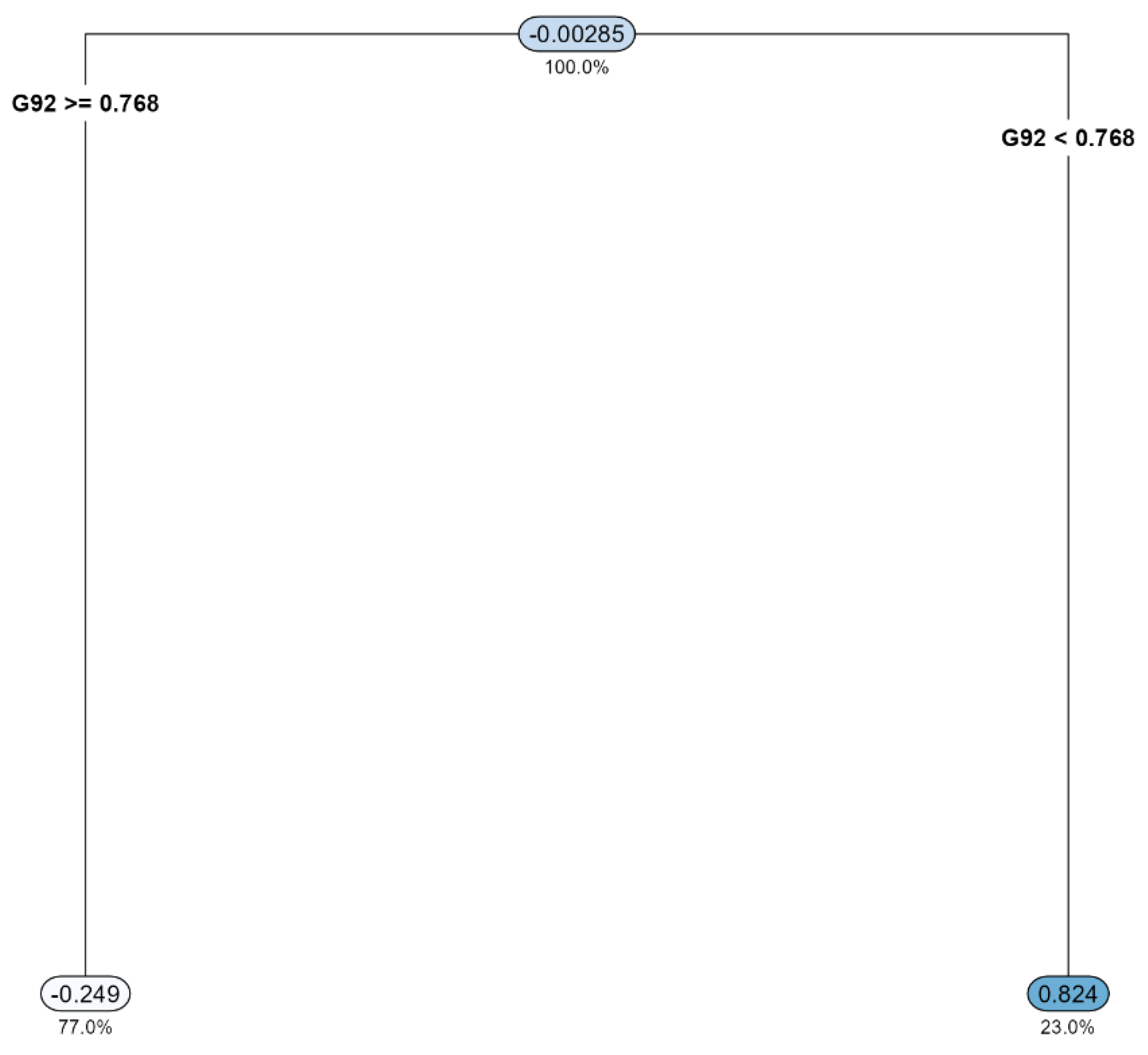
3.3. Data Mining: Decision Tree
3.4. Data Mining: Random Forest
3.5. Data Mining: Gradient Boosting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Garrett, W.S.; Gordon, J.I.; Glimcher, L.H. Homeostasis and Inflammation in the Intestine. Cell 2010, 140, 859–870. [Google Scholar] [CrossRef]
- Cox, L.M.; Blaser, M.J. Antibiotics in Early Life and Obesity. Nat. Rev. Endocrinol. 2015, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the Intestinal Microbiota during a Critical Developmental Window Has Lasting Metabolic Consequences. Cell 2014, 158, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Koh, H.; Kurtz, Z.D.; Battaglia, T.; PeBenito, A.; Li, H.; Nazzal, L.; Blaser, M.J. Oxalobacter Formigenes-Associated Host Features and Microbial Community Structures Examined Using the American Gut Project. Microbiome 2017, 5, 108. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current Understanding of the Human Microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Zhang, X.S.; Li, J.; Krautkramer, K.A.; Badri, M.; Battaglia, T.; Borbet, T.C.; Koh, H.; Ng, S.; Sibley, R.A.; Li, Y.; et al. Antibiotic-Induced Acceleration of Type 1 Diabetes Alters Maturation of Innate Intestinal Immunity. Elife 2018, 7, e37816. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, G.; Ianiro, G.; Ahern, A.; Carbone, C.; Temko, A.; Claesson, M.J.; Gasbarrini, A.; Tortora, G. Gut Microbiome, Big Data and Machine Learning to Promote Precision Medicine for Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 635–648. [Google Scholar] [CrossRef]
- Sharma, A.; Im, S.H. Special Issue on the Human Microbiome: From Symbiosis to Therapy. Exp. Mol. Med. 2020, 52, 1361–1363. [Google Scholar] [CrossRef]
- Zhao, N.; Khamash, D.F.; Koh, H.; Voskertchian, A.; Egbert, E.; Mongodin, E.F.; White, J.R.; Hittle, L.; Colantuoni, E.; Milstone, A.M. Low Diversity in Nasal Microbiome Associated with Staphylococcus aureus Colonization and Bloodstream Infections in Hospitalized Neonates. Open Forum Infect. Dis. 2021, 8, ofab475. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- VanEvery, H.; Franzosa, E.A.; Nguyen, L.H.; Huttenhower, C. Microbiome Epidemiology and Association Studies in Human Health. Nat. Rev. Genet. 2023, 24, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Microbiome, Metagenomics, and High-Dimensional Compositional Data Analysis. Annu. Rev. Stat. Its Appl. 2015, 2, 73–94. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.H.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC Press: Boca Raton, FL, USA, 1984. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Hastie, T.; Friedman, J.H.; Tibshirani, R. The Elements of Statistical Learning, 2nd ed.; Springer Series in Statistics; Springer: New York, NY, USA, 2009; ISBN 978-0-387-84857-0. [Google Scholar]
- Chang, H.X.; Haudenshield, J.S.; Bowen, C.R.; Hartman, G.L. Metagenome-Wide Association Study and Machine Learning Prediction of Bulk Soil Microbiome and Crop Productivity. Front. Microbiol. 2017, 8, 519. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Steinert, G.; Nielsen, S.; Hardoim, C.C.; Wu, Y.C.; McCormack, G.P.; López-Legentil, S.; Marchant, R.; Webster, N.; Thomas, T.; et al. Predicting the HMA-LMA Status in Marine Sponges by Machine Learning. Front. Microbiol. 2017, 8, 752. [Google Scholar] [CrossRef] [PubMed]
- Hernández Medina, R.; Kutuzova, S.; Nielsen, K.N.; Johansen, J.; Hansen, L.H.; Nielsen, M.; Rasmussen, S. Machine Learning and Deep Learning Applications in Microbiome Research. ISME Commun. 2022, 2, 98. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Moon, J.; Chisina, C.; Kang, B.; Park, T.; Koh, H. MiCloud: A Unified Web Platform for Comprehensive Microbiome Data Analysis. PLoS ONE 2022, 17, e0272354. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Koh, H.; Gu, W.; Kang, B. Integrative Web Cloud Computing and Analytics Using MiPair for Design-Based Comparative Analysis with Paired Microbiome Data. Sci. Rep. 2022, 12, 20465. [Google Scholar] [CrossRef]
- Gu, W.; Koh, H.; Jang, H.; Lee, B.; Kang, B. MiSurv: An Integrative Web Cloud Platform for User-Friendly Microbiome Data Analysis with Survival Responses. Microbiol. Spectr. 2023, 10, e05059-22. [Google Scholar] [CrossRef]
- Jang, H.; Park, S.; Koh, H. Comprehensive Microbiome Causal Mediation Analysis Using MiMed on User-Friendly Web Interfaces. Biol. Methods Protoc. 2023, 8, bpad023. [Google Scholar] [CrossRef]
- Huse, S.M.; Mark Welch, D.B.; Voorhis, A.; Shipunova, A.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. VAMPS: A Website for Visualization and Analysis of Microbial Population Structures. BMC Bioinform. 2014, 15, 41. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S rRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Shi, W.; Qi, H.; Sun, Q.; Fan, G.; Liu, S.; Wang, J.; Zhu, B.; Liu, H.; Zhao, F.; Wang, X.; et al. gcMeta: A Global Catalogue of Metagenomics Platform to Support the Archiving, Standardization and Analysis of Microbiome Data. Nucleic Acids Res. 2019, 47, D637–D648. [Google Scholar] [CrossRef]
- Jin, B.T.; Xu, F.; Ng, R.T.; Hogg, J.C. Mian: Interactive Web-Based Microbiome Data Table Visualization and Machine Learning Platform. Bioinformatics 2022, 38, 1176–1178. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.; Liou, D.; Dommer, J.; MacMenamin, P.; Quiñones, M.; Misner, I.; Oler, A.J.; Wan, J.; Kim, L.; McCarthy, M.C.; et al. Nephele: A Cloud Platform for Simplified, Standardized and Reproducible Microbiome Data Analysis. Bioinformatics 2018, 34, 1411–1413. [Google Scholar] [CrossRef]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, Web-Enabled Microbiome Meta-Analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Mitchell, K.; Ronas, J.; Dao, C.; Freise, A.C.; Mangul, S.; Shapiro, C.; Moberg-Parker, J. PUMAA: A Platform for Accessible Microbiome Analysis in the Undergraduate Classroom. Front. Microbiol. 2020, 11, 584699. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Koh, H.; Patatanian, M.; Reyes-Caballero, H.; Zhao, N.; Meinert, J.; Holbrook, J.T.; Leinbach, L.I.; Biswal, S. The Mediating Roles of the Oral Microbiome in Saliva and Subgingival Sites between E-Cigarette Smoking and Gingival Inflammation. BMC Microbiol. 2023, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Fox, G.E. Phylogenetic Structure of the Prokaryotic Domain: The Primary Kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a Natural System of Organisms: Proposal for the Domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New Insights into Human Nostril Microbiome from the Expanded Human Oral Microbiome Database (eHOMD): A Resource for the Microbiome of the Human Aerodigestive Tract. mSystems 2018, 3, e00187-18. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data. J. R. Stat. Soc. Ser. B Methodol. 1982, 44, 139–160. [Google Scholar] [CrossRef]
- Sanders, H.L. Marine Benthic Diversity: A Comparative Study. Am. Nat. 1968, 102, 243–282. [Google Scholar] [CrossRef]
- Manduchi, E.; Fu, W.; Romano, J.D.; Ruberto, S.; Moore, J.H. Embedding Covariate Adjustments in Tree-Based Automated Machine Learning for Biomedical Big Data Analyses. BMC Bioinform. 2020, 21, 430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A Guide from Sampling to Data Analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Lee, S. A Unified Approach to Interpreting Model Predictions. In Proceedings of the Advances in Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 4765–4774. [Google Scholar]
- Tukey, J.W. The Teaching of Concrete Mathematics. Am. Math. Mon. 1958, 65, 1–9. [Google Scholar] [CrossRef]
- Su, Q.; Liu, Q.; Lau, R.I.; Zhang, J.; Xu, Z.; Yeoh, Y.K.; Leung, T.W.H.; Tang, W.; Zhang, L.; Liang, J.Q.Y.; et al. Faecal Microbiome-Based Machine Learning for Multi-Class Disease Diagnosis. Nat. Comm. 2022, 13, 6818. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, F.; Siarheyeu, R.; Alqassem, I.; Henschel, A.; Pileggi, G.; Meiser, A. Microbiome-Based Disease Prediction with Multimodal Variational Information Bottlenecks. PLoS Comput. Biol. 2022, 18, e1010050. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Decision Tree | Random Forest * | Gradient Boosting * |
|---|---|---|---|
| Interpretability | Very easy | Easy | Easy |
| Prediction accuracy | Moderate | Very accurate | Very accurate |
| Computational speed | Very fast | Moderate | Slow |
| Platform | Main Facility | Output Variable | Covariate-Adjustment | Study Design |
|---|---|---|---|---|
| MiCloud | Association testing | Binary/continuous | Yes | Cross-sectional, Family-based, repeated measures |
| MiPair | Paired analysis | Continuous | No | Paired/Block |
| MiSurv | Survival analysis | Time-to-event | Yes | Follow-up |
| MiMed | Mediation analysis | Binary/continuous | Yes | Cross-sectional |
| MiTree | Prediction modeling | Binary/continuous | Yes | Cross-sectional |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Koh, H. MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods. Microorganisms 2023, 11, 2816. https://doi.org/10.3390/microorganisms11112816
Kim J, Koh H. MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods. Microorganisms. 2023; 11(11):2816. https://doi.org/10.3390/microorganisms11112816
Chicago/Turabian StyleKim, Jihun, and Hyunwook Koh. 2023. "MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods" Microorganisms 11, no. 11: 2816. https://doi.org/10.3390/microorganisms11112816
APA StyleKim, J., & Koh, H. (2023). MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods. Microorganisms, 11(11), 2816. https://doi.org/10.3390/microorganisms11112816