
Gut Microbiota and Metabolites May Play a Crucial Role in Sea Cucumber Apostichopus japonicus Aestivation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals, Grouping, and Feeding Conditions
2.2. Analysis of Weight Change
2.3. Microbial Diversity Analysis
2.3.1. Sequencing
2.3.2. Data Analysis
2.4. Metabolomic Analysis
2.4.1. Metabolite Extraction
2.4.2. UHPLC-MS/MS Analysis
2.4.3. Data Processing, Metabolite Identification, and Data Analysis
2.5. Correlation Analysis
3. Results
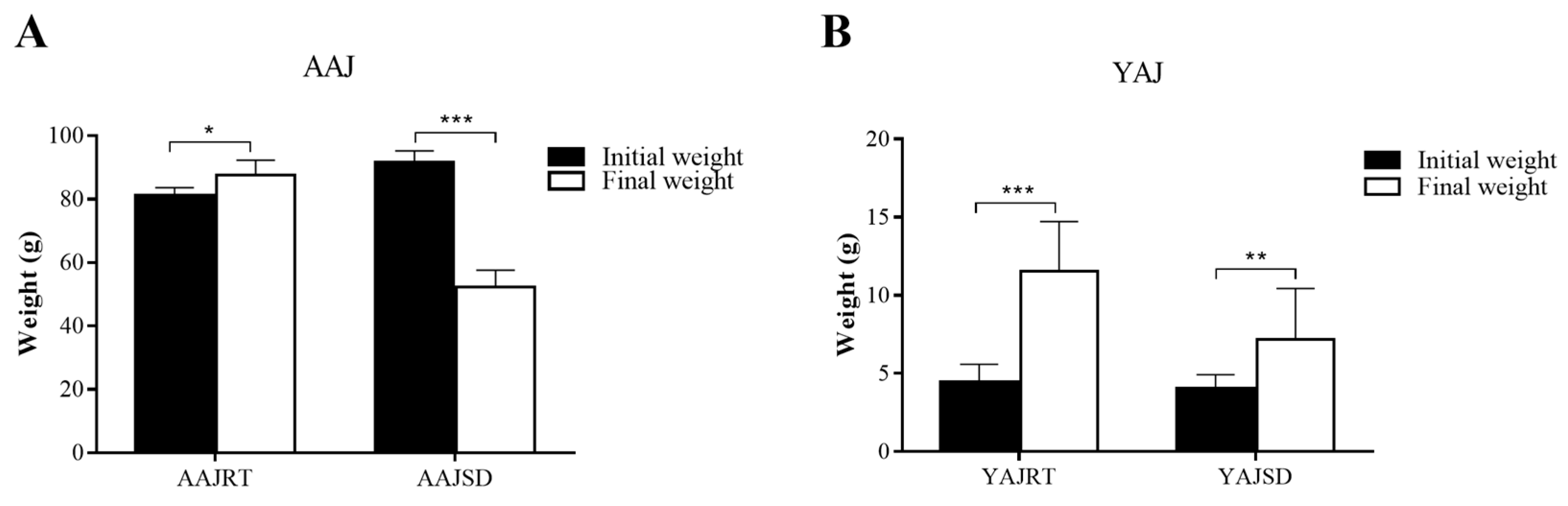
3.1. The Effect of High Temperature on the Growth of Adult and Young A. Japonicus
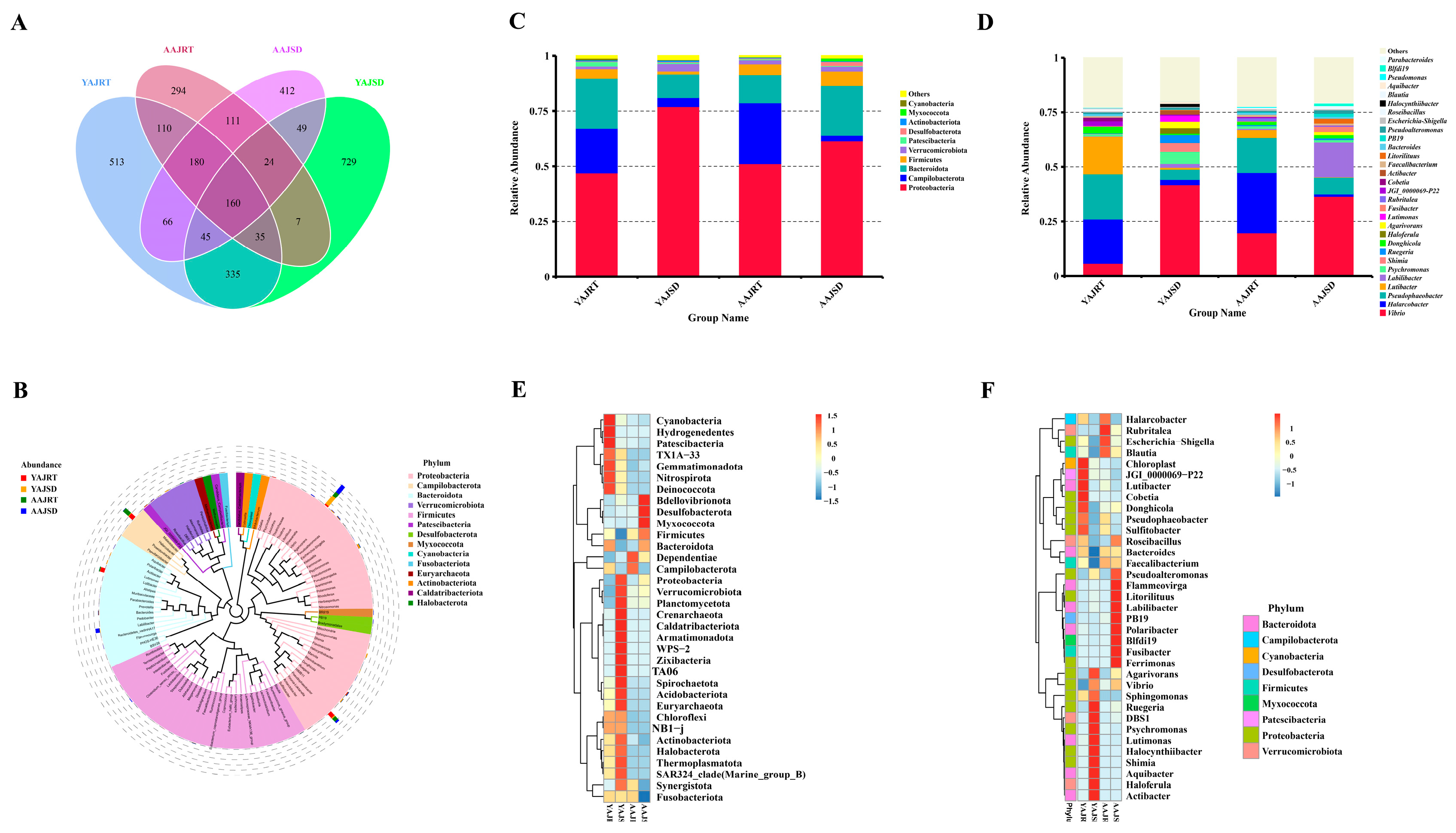
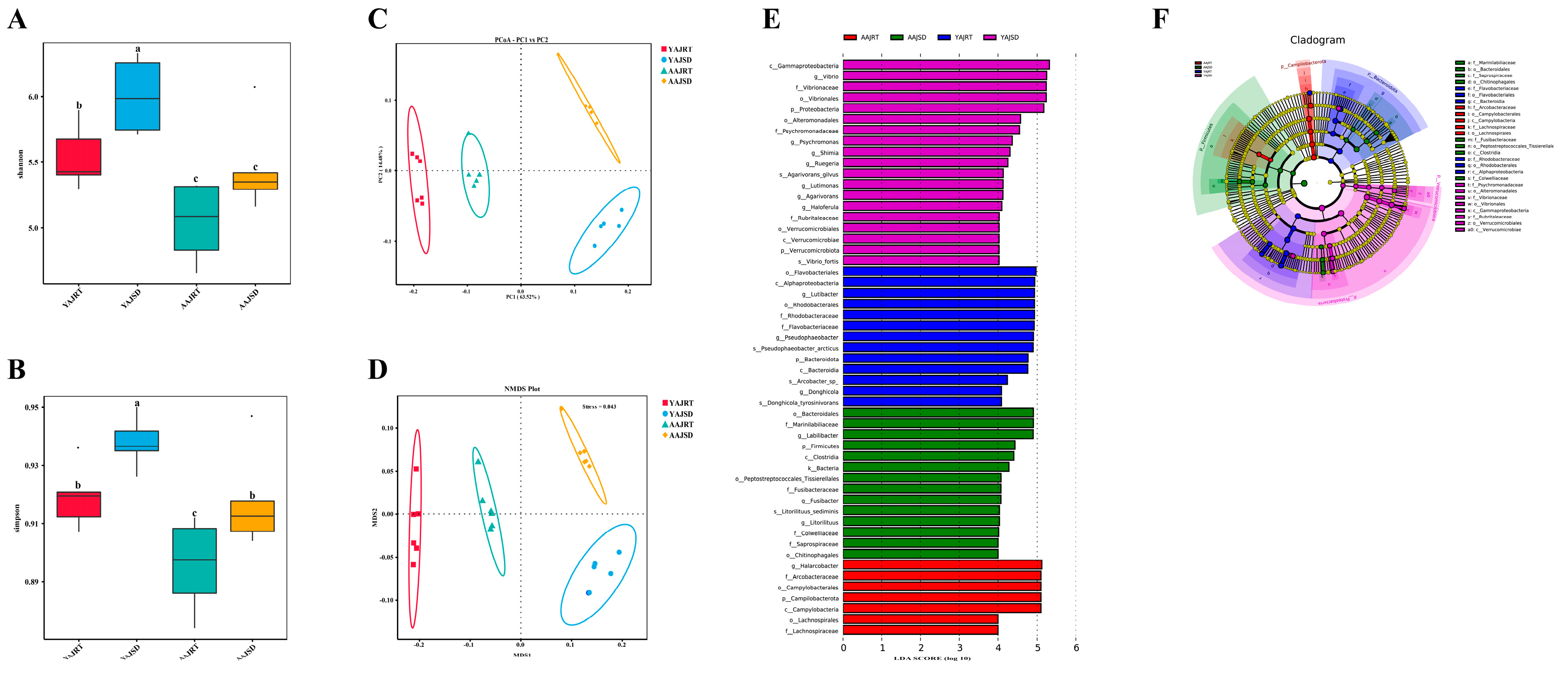
3.2. Microbial Diversity Analysis
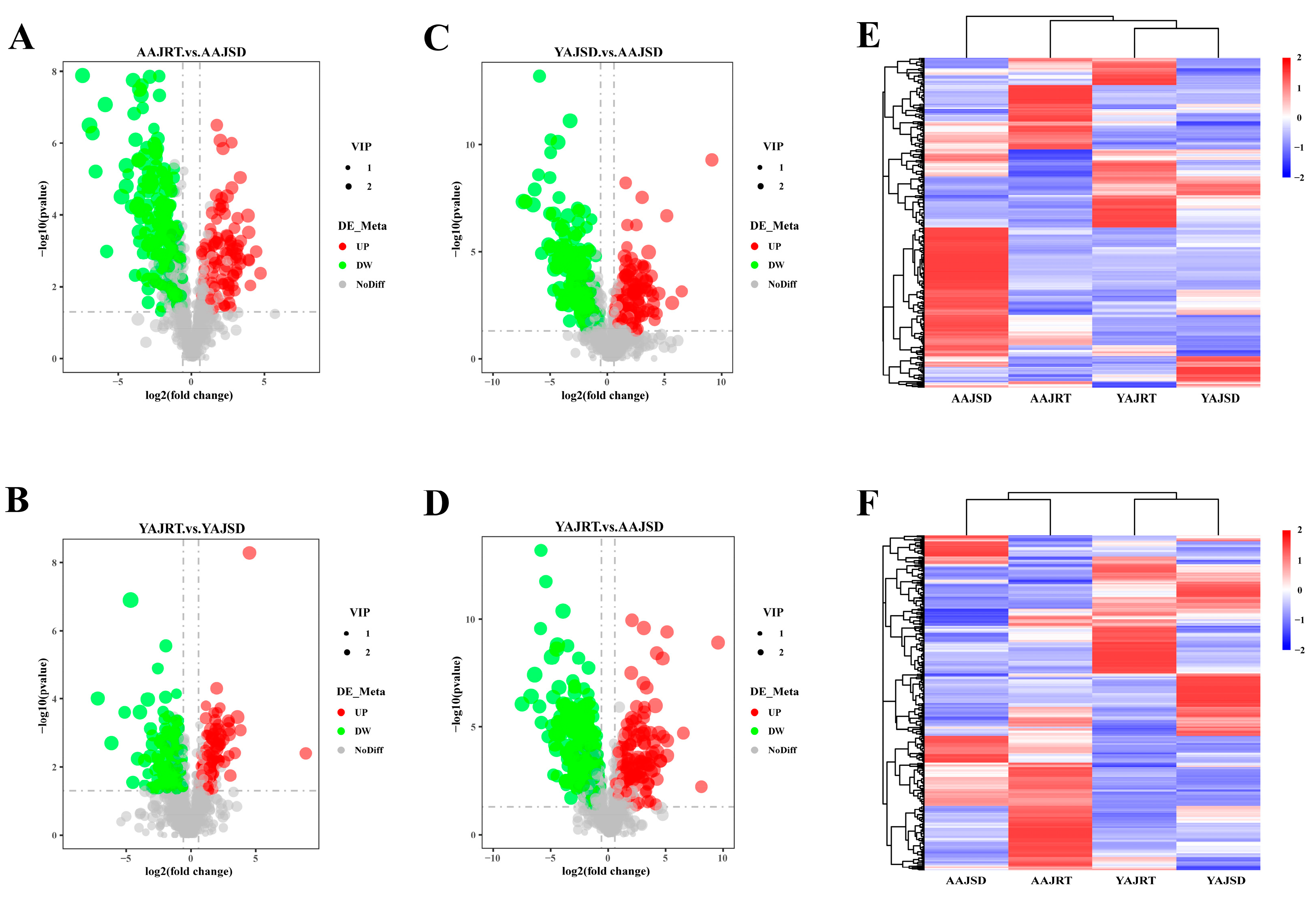
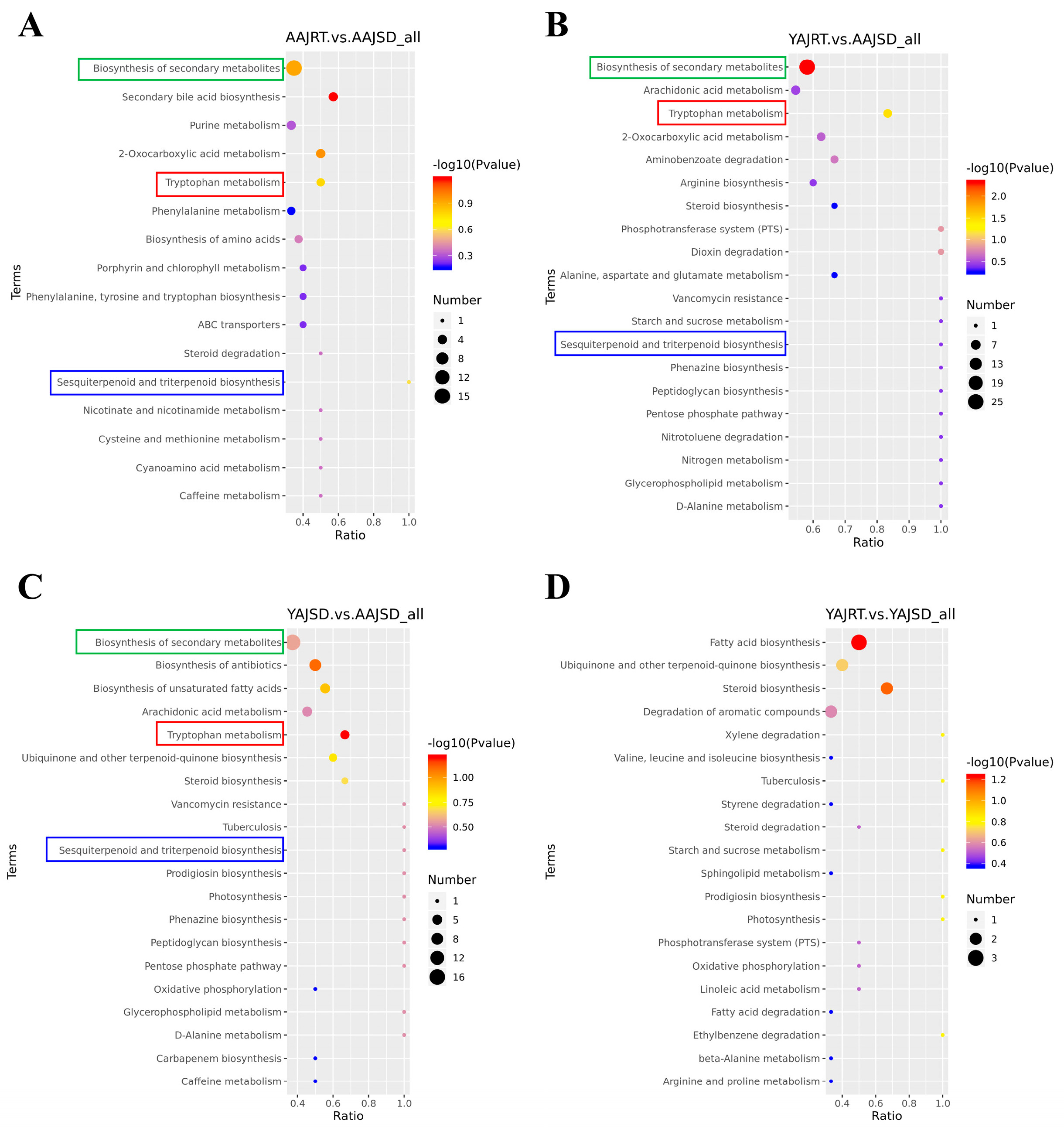
3.3. Metabolomics Analysis
3.4. Correlation between Differentially Abundant Microbes and Differential Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.S.; Sun, J.C.; Ru, X.S.; Zhang, C.Y.; Xing, L.L.; Sun, L.N.; Lin, C.G.; Liu, S.L.; Zhang, L.B. Current advances and technological prospects of the sea cucumber seed industry in China. Sci. Mar. 2020, 44, 2–9. [Google Scholar]
- Mohsen, M.; Yang, H. Sea Cucumbers: Aquaculture, Biology and Ecology; American Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Li, Y.; Ding, Y.; Pan, L. Effects of increasing temperature and aestivation on biogenic amines, signal transduction pathways and metabolic enzyme activities in the sea cucumber (Apostichopus japonicus). Mar. Biol. 2022, 169, 9. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, X.; Lu, Z.; Huang, R.; Tran, N.T.; Wu, J.; Yang, F.; Ge, H.; Zhong, C.; Sun, Q.; et al. Transcriptome and Metabolome Analyses of Sea Cucumbers Apostichopus japonicus in Southern China During the Summer Aestivation Period. J. Ocean Univ. China 2021, 20, 198–212. [Google Scholar] [CrossRef]
- Gao, L.; Yuan, Z.; Li, Y.; Ma, Z. Genome-wide comparative analysis of DNAJ genes and their co-expression patterns with HSP70s in aestivation of the sea cucumber Apostichopus japonicus. Funct. Integr. Genom. 2022, 22, 317–330. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Chen, M.; Storey, K.B.; Wang, T. A potential antiapoptotic regulation: The interaction of heat shock protein 70 and apoptosis-inducing factor mitochondrial 1 during heat stress and aestivation in sea cucumber. J. Exp. Zool. Part A 2018, 329, 103–111. [Google Scholar] [CrossRef]
- Chen, M.; Wang, S.; Li, X.; Storey, K.B.; Zhang, X. The potential contribution of miRNA-200-3p to the fatty acid metabolism by regulating AjEHHADH during aestivation in sea cucumber. PeerJ Comput. Sci. 2018, 6, e5703. [Google Scholar]
- Yang, Y.; Zheng, Y.; Sun, L.; Chen, M. Genome-Wide DNA Methylation Signatures of Sea Cucumber Apostichopus japonicus during Environmental Induced Aestivation. Genes 2020, 11, 1020. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, A.; Storey, K.B. Comparative phosphoproteomic analysis of intestinal phosphorylated proteins in active versus aestivating sea cucumbers. J. Proteom. 2016, 135, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, R.; Xun, X.; Wang, J.; Bao, L.; Thimmappa, R.; Ding, J.; Jiang, J.; Zhang, L.; Li, T.; et al. Sea cucumber genome provides insights into saponin biosynthesis and aestivation regulation. Cell Discov. 2018, 4, 29. [Google Scholar] [CrossRef]
- Sen, P.; Molinero-Perez, A.; O’Riordan, K.J.; McCafferty, C.P.; O’Halloran, K.D.; Cryan, J.F. Microbiota and sleep: Awakening the gut feeling. Trends Mol. Med. 2021, 27, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hao, Y.; Fan, F.; Zhang, B. The Role of Microbiome in Insomnia, Circadian Disturbance and Depression. Front. Psychiatry 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Matenchuk, B.A.; Mandhane, P.J.; Kozyrskyj, A.L. Sleep, circadian rhythm, and gut microbiota. Sleep Med. Rev. 2020, 53, 101340. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Miyoshi, C.; Obana, N.; Yajima, K.; Hotta-Hirashima, N.; Ikkyu, A.; Kanno, S.; Soga, T.; Fukuda, S.; Yanagisawa, M. Gut microbiota depletion by chronic antibiotic treatment alters the sleep/wake architecture and sleep EEG power spectra in mice. Sci. Rep. 2020, 10, 19554. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Bhat, S.; Li, W.; Wei, Y.; Kui, H.; Fu, X.; Gui, H.; Wei, C.; Li, F. Gut digestion of earthworms significantly attenuates cell-free and -associated antibiotic resistance genes in excess activated sludge by affecting bacterial profiles. Sci. Total. Environ. 2019, 691, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Sheik, C.S.; Krumholz, L.R.; Najar, F.Z.; Roe, B.A.; Elshahed, M.S. Comparison of Species Richness Estimates Obtained Using Nearly Complete Fragments and Simulated Pyrosequencing-Generated Fragments in 16S rRNA Gene-Based Environmental Surveys. Appl. Environ. Microbiol. 2009, 75, 5227–5236. [Google Scholar] [CrossRef]
- Li, Y.L.; Wang, R.J.; Xun, X.G.; Wang, J.; Bao, L.S.; Thimmappa, R.; Ding, J.; Jiang, J.W.; Zhang, L.H.; Li, T.Q.; et al. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Li, M.; Shao, D.; Zhou, J.; Gu, J.; Qin, J.; Chen, W.; Wei, W. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chin. J. Cancer Res. 2020, 32, 755–767. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Sellick, C.A.; Hansen, R.; Stephens, G.M.; Goodacre, R.; Dickson, A.J. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat. Protoc. 2011, 6, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yang, H.; Wang, L.; Zhou, Y.; Zhang, T.; Liu, Y. Effects of aestivation on the energy budget of sea cucumber Apostichopus japonicus (Selenka) (Echinodermata: Holothuroidea). Acta Ecol. Sin. 2007, 27, 3155–3161. [Google Scholar] [CrossRef]
- Smith, R.P.; Easson, C.; Lyle, S.M.; Kapoor, R.; Donnelly, C.P.; Davidson, E.J.; Parikh, E.; Lopez, J.V.; Tartar, J.L. Gut microbiome diversity is associated with sleep physiology in humans. PLoS ONE 2019, 14, e0222394. [Google Scholar] [CrossRef]
- Meloni, M.; Figorilli, M.; Carta, M.; Tamburrino, L.; Cannas, A.; Sanna, F.; Defazio, G.; Puligheddu, M. Preliminary finding of a randomized, double-blind, placebo-controlled, crossover study to evaluate the safety and efficacy of 5-hydroxytryptophan on REM sleep behavior disorder in Parkinson’s disease. Sleep. Breath 2022, 26, 1023–1031. [Google Scholar] [CrossRef]
- Szczepanik, M. Melatonin and its influence on immune system. J. Physiol. Pharmacol. 2007, 58, 115–124. [Google Scholar]
- Gehrke, S.; Rice, S.; Stefanoni, D.; Wilkerson, R.B.; Nemkov, T.; Reisz, J.A.; Hansen, K.C.; Lucas, A.; Cabrales, P.; Drew, K.; et al. Red Blood Cell Metabolic Responses to Torpor and Arousal in the Hibernator Arctic Ground Squirrel. J. Proteome Res. 2019, 18, 1827–1841. [Google Scholar] [CrossRef]
- Hiong, K.C.; Ip, Y.K.; Wong, W.P.; Chew, S.F. Differential Gene Expression in the Brain of the African Lungfish, Protopterus annectens, after Six Days or Six Months of Aestivation in Air. PLoS ONE 2013, 8, e71205. [Google Scholar] [CrossRef]
- Popova, N.; Voronova, I.; Kulikov, A. Involvement of brain tryptophan hydroxylase in the mechanism of hibernation. Pharmacol. Biochem. Behav. 1993, 46, 9–13. [Google Scholar] [CrossRef]
- Tricklebank, M.D. Serotonin and sleep. In The Serotonin System; American Academic Press: London, UK, 2019; pp. 181–192. [Google Scholar]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276, Erratum in 2015, 163, 258. [Google Scholar] [CrossRef]
- Wu, W.; Kong, Q.; Tian, P.; Zhai, Q.; Wang, G.; Liu, X.; Zhao, J.; Zhang, H.; Lee, Y.K.; Chen, W. Targeting Gut Microbiota Dysbiosis: Potential Intervention Strategies for Neurological Disorders. Engineering 2020, 6, 415–423. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Wang, H.; Denou, E.; Ghia, J.-E.; Rossi, L.; Fontes, M.E.; Bernier, S.P.; Shajib, M.S.; Banskota, S.; Collins, S.M.; et al. Modulation of Gut Microbiota Composition by Serotonin Signaling Influences Intestinal Immune Response and Susceptibility to Colitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 709–728. [Google Scholar] [CrossRef]
- Gheorghe, C.E.; Martin, J.A.; Manriquez, F.V.; Dinan, T.G.; Cryan, J.F.; Clarke, G. Focus on the essentials: Tryptophan metabolism and the microbiome-gut-brain axis. Curr. Opin. Pharmacol. 2019, 48, 137–145. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.-L.; Farzi, A.; Zhu, W.-Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in silico Analysis. Front. Mol. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Name | FC | p-Value | VIP | Up or Down |
|---|---|---|---|---|---|
| AAJSD vs. AAJRT | 5-hydroxytryptophan | 3.731 | 0.000309 | 1.617 | Up |
| AAJSD vs. YAJSD | 5-hydroxytryptophan | 2.695 | 0.009288 | 1.103 | Up |
| AAJSD vs. YAJRT | 5-hydroxytryptophan | 2.968 | 0.000472 | 1.504 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.-H.; Yang, B.-T.; Hu, R.-G.; Zhang, P.; Gu, M.; Cong, W. Gut Microbiota and Metabolites May Play a Crucial Role in Sea Cucumber Apostichopus japonicus Aestivation. Microorganisms 2023, 11, 416. https://doi.org/10.3390/microorganisms11020416
Kang Y-H, Yang B-T, Hu R-G, Zhang P, Gu M, Cong W. Gut Microbiota and Metabolites May Play a Crucial Role in Sea Cucumber Apostichopus japonicus Aestivation. Microorganisms. 2023; 11(2):416. https://doi.org/10.3390/microorganisms11020416
Chicago/Turabian StyleKang, Yuan-Huan, Bin-Tong Yang, Ren-Ge Hu, Peng Zhang, Min Gu, and Wei Cong. 2023. "Gut Microbiota and Metabolites May Play a Crucial Role in Sea Cucumber Apostichopus japonicus Aestivation" Microorganisms 11, no. 2: 416. https://doi.org/10.3390/microorganisms11020416