

Metagenomics-Based Analysis of Candidate Lactate Utilizers from the Rumen of Beef Cattle



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Rumen Culture Experiments
2.2. Microbial Genomic DNA Purification and PCR Amplification of the 16S rRNA Gene
2.3. Bioinformatics Pipeline for 16S rRNA Gene-Based Composition Analysis
2.4. Metagenomics Analysis
2.5. Availability and Accession of Next Generation Sequencing Data
3. Results
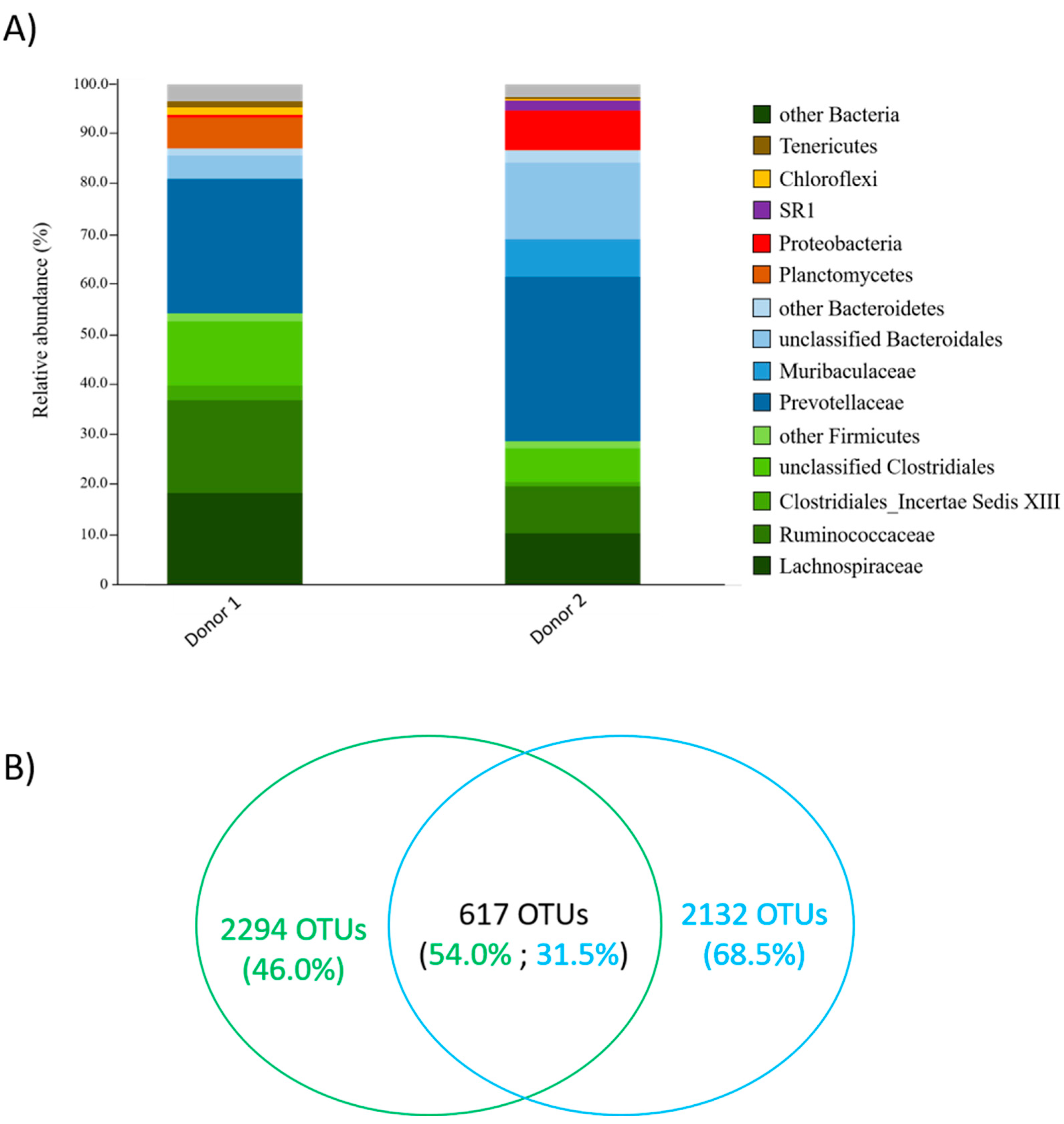
3.1. Identification of Candidate Lactate Utilizers from Rumen Fluid
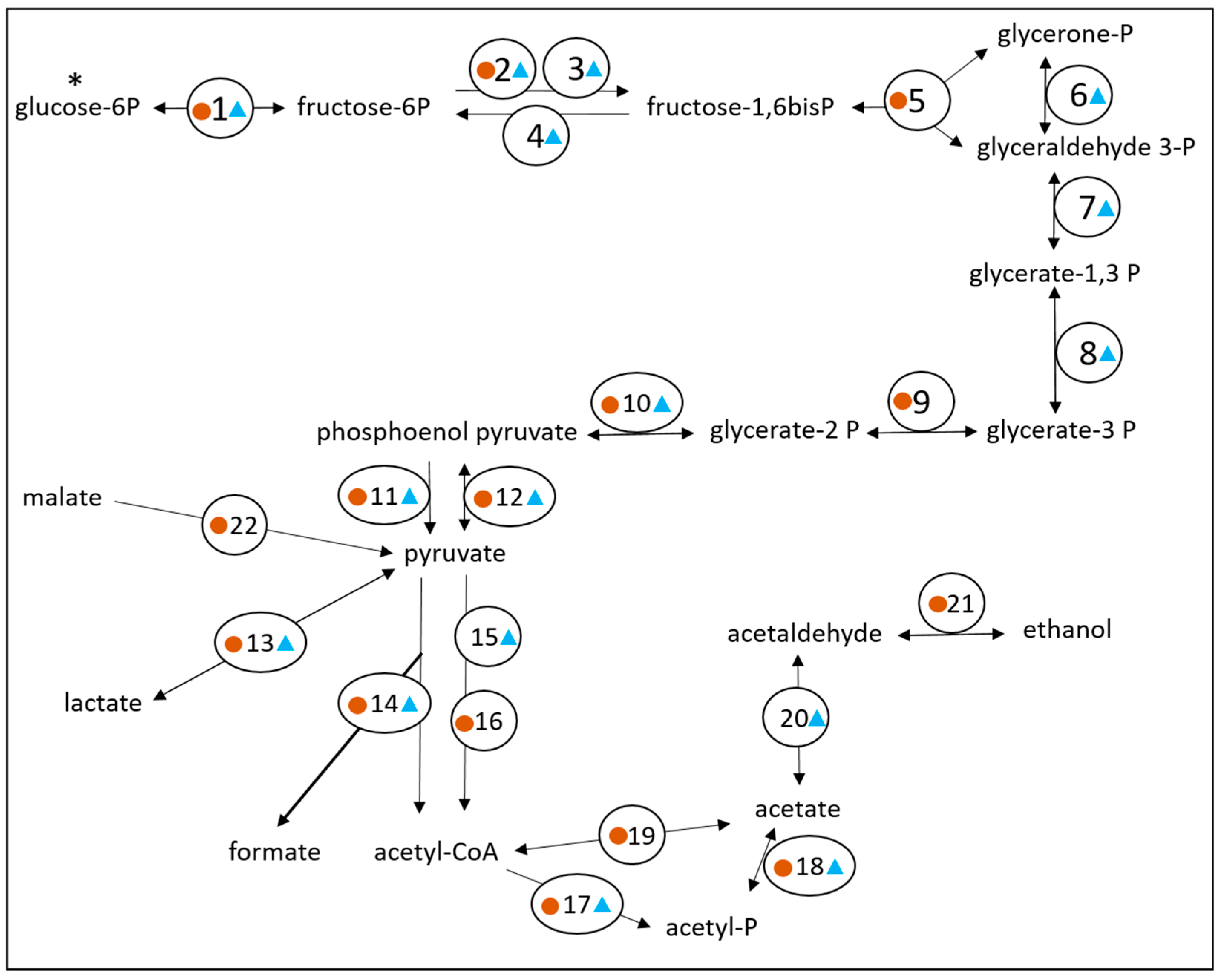
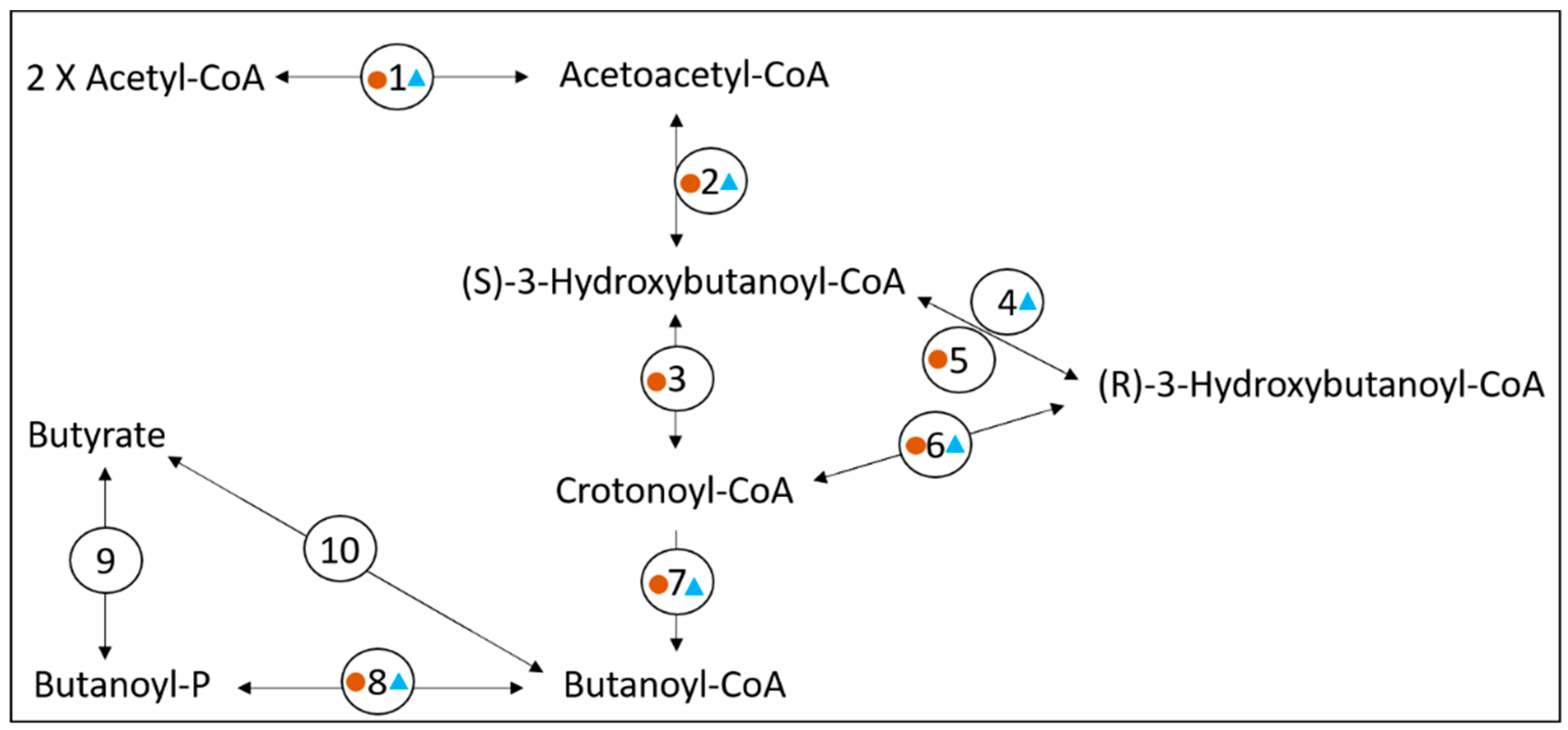
3.2. Exploring the Metabolic Potential of OTU Bt-01708_Bf
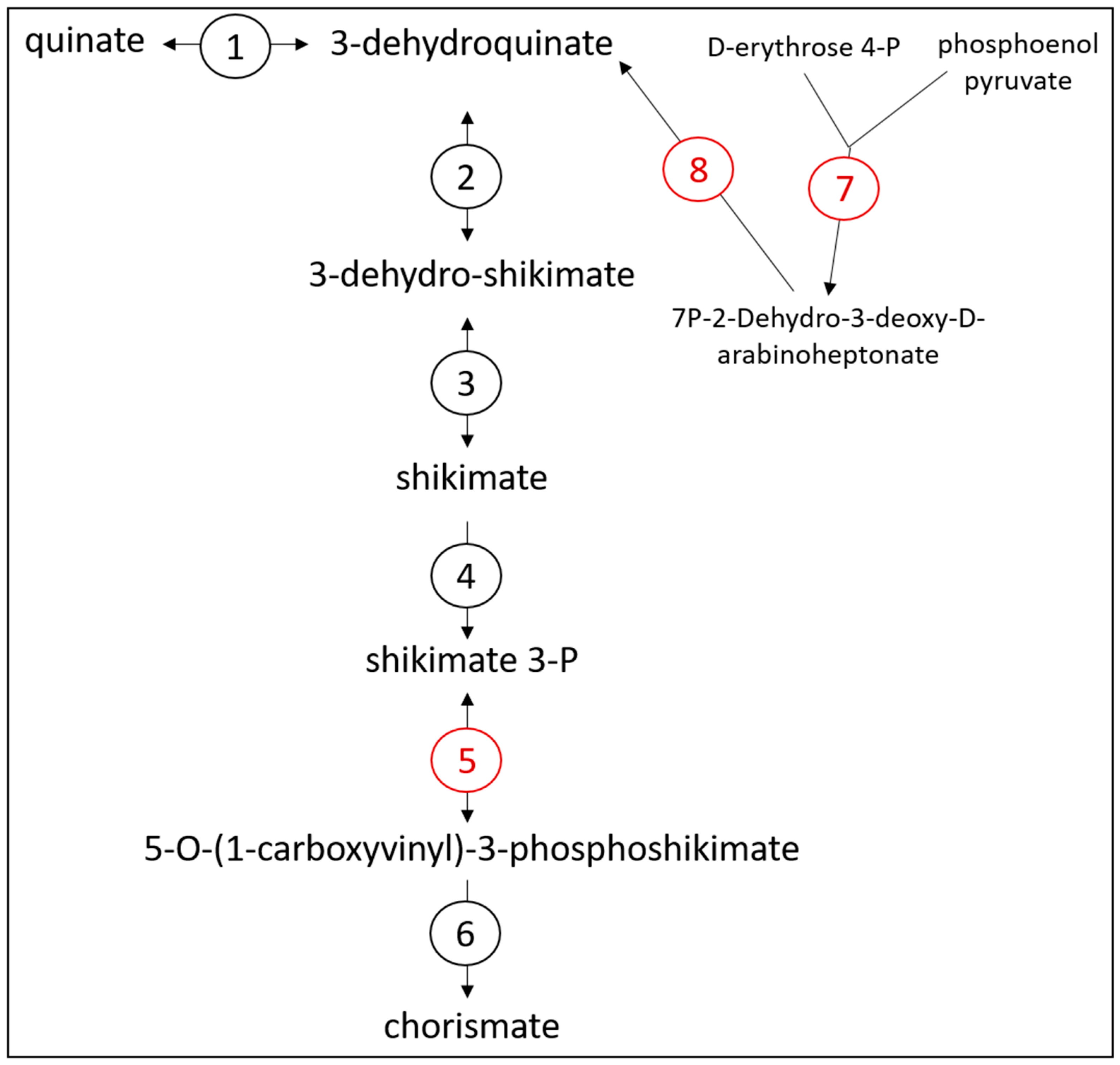
3.3. Exploring the Metabolic Potential of OTU Bt-01899_Ap
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McSweeney, C.; Mackie, R. Microorganisms and Ruminant Digestion: State of Knowledge, Trends and Future Prospects. Background Study Paper No. 61; Food and Agriculture Organization of the United Nations: Rome, Italy, 2012. [Google Scholar]
- Hackmann, T.J.; Spain, J.N. Invited Review: Ruminant ecology and evolution: Perspectives useful to ruminant livestock research and production. J. Dairy Sci. 2010, 93, 1320–1334. [Google Scholar] [CrossRef]
- Thornton, P.K. Livestock production: Recent trends, future prospects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2853–2867. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.T.; Macfarlane, S. Factors affecting fermentation reactions in the large bowel. Proc. Nutr. Soc. 1993, 52, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, R.R. Evolutionary steps of ecophysiological adaptation and diversification of ruminants: A comparative view of their digestive system. Oecologia 1989, 78, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Goldschmidt, F.; Lilja, E.E.; Ackermann, M. Metabolic specialization and the assembly of microbial communities. ISME J. 2012, 6, 1985–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef] [Green Version]
- Weimer, P.J.; Russell, J.B.; Muck, R.E. Lessons from the cow: What the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass. Bioresour. Technol. 2009, 100, 5323–5331. [Google Scholar] [CrossRef]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaizier, J.C.; Mulligan, F.J.; Neville, E.W.; Guan, L.L.; Steele, M.A.; Penner, G.B. Invited review: Effect of subacute ruminal acidosis on gut health of dairy cows. J. Dairy Sci. 2022, 105, 7141–7160. [Google Scholar] [CrossRef]
- Krause, K.M.; Oetzel, G.R. Understanding and preventing subacute ruminal acidosis in dairy herds: A review. Anim. Feed Sci Technol. 2006, 126, 215–236. [Google Scholar] [CrossRef]
- Nagaraja, T.G.; Titgemeyer, E.C. Ruminal acidosis in beef cattle: The current microbiological and nutritional outlook. J. Dairy Sci. 2007, 90, E17–E38. [Google Scholar] [CrossRef] [Green Version]
- Slyter, L.L. Influence of acidosis on rumen function. J. Anim. Sci. 1976, 43, 910–929. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Li, S.; Danscher, A.M.; Derakshani, H.; Andersen, P.H.; Khafipour, E. Changes in microbiota in rumen digesta and feces due to a grain-based subacute ruminal acidosis (SARA) challenge. Microb. Ecol. 2017, 74, 485–495. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Li, S.; Tun, H.M.; Khafipour, E. Nutritional models of experimentally-induced subacute ruminal acidosis (SARA) differ in their impact on rumen and hindgut bacterial communities in dairy cows. Front. Microbiol. 2016, 7, 2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, J.C.; Luan, S.; Cardoso, F.C.; Derakhshani, H.; Khafipour, E.; Loor, J.J. Induction of subacute ruminal acidosis affects the ruminal microbiome and epithelium. Front. Microbiol. 2016, 7, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Liu, J.; Mao, S. Impact of subacute ruminal acidosis (SARA) on ruminal microbiome, lipopolysaccharide and bioamine and rumen epithelial health of dairy cows. J. Dairy Vet. Sci. 2017, 1, 1. [Google Scholar]
- Khafipour, E.; Li, S.; Plaizier, J.C.; Krause, D.O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 2009, 75, 7115–7124. [Google Scholar] [CrossRef] [Green Version]
- Khafipour, E.; Li, S.; Tun, H.M.; Derakkhshani, H.; Moossavi, S.; Plaizier, J.C. Effects of grain feeding on microbiota in the digestive tract of cattle. Anim. Front. 2016, 6, 13–19. [Google Scholar] [CrossRef]
- Mao, S.Y.; Zhang, R.; Wang, D.; Zhu, W. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 2013, 24, 12–19. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, E.; Neves, A.L.; Song, Y.; Guan, L.L. The role of the gut microbiome in cattle production and health: Driver or passenger? Annu. Rev. Anim. Biosci. 2020, 8, 199–220. [Google Scholar] [CrossRef] [Green Version]
- Brede, M.; Orton, T.; Pinior, B.; Roch, F.F.; Dzieciol, M.; Zwirzitz, B.; Wagner, M.; Breves, G.; Wetzels, S.U. PacBiol. and Illumina MiSeq amplicon sequencing confirm full recovery of the bacterial community after subacute ruminal acidosis challenge in the Rusitec system. Front. Microbiol. 2020, 11, 1813. [Google Scholar] [CrossRef] [PubMed]
- Ogunade, I.; Pech-Cervantes, A.; Schweickart, H. Metatranscriptomic analysis of sub-acute ruminal acidosis in beef cattle. Animals 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tun, H.M.; Li, S.; Yoon, I.; Meale, S.J.; Azevedo, P.A.; Khafipour, E.; Plaizier, J.C. Saccharomyces cerevisiae fermentation products (SCFP) stabilize the ruminal microbiota of lactating dairy cows during periods of a depressed rumen pH. BMC Vet. Res. 2020, 16, 237. [Google Scholar] [CrossRef]
- Fernando, S.C.; Purvis II, H.T.; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; Desilva, U.J. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, R.; Altman, E.; Eiteman, M.A. Lactate and acrylate metabolism by Megasphaera elsdenii under batch and steady-state conditions. Appl. Environ. Microbiol. 2012, 78, 8564–8570. [Google Scholar] [CrossRef] [Green Version]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 2013, 79, 3744–3755. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.; Benedito, J.L.; Abuelo, A.; Castillo, C. Ruminal acidosis in feedlot: From aetiology to prevention. Sci. World J. 2014, 2014, 702572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, N.; Vanzant, E.; Harmon, D.; McLeod, K. Direct-fed microbials containing lactate-producing bacteria influence ruminal fermentation but not lactate utilization in steers fed a high-concentrate diet. J. Anim. Sci. 2015, 93, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Creevey, C.J.; Kelly, W.J.; Henderson, G.; Leahy, S.C. Determining the culturability of the rumen bacterial microbiome. Microb. Biotechnol. 2014, 7, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Huws, S.A.; Creevey, C.J.; Oyama, L.B.; Mizrahi, I.; Denman, S.E.; Popova, M.; Muñoz-Tamayo, R.; Forano, E.; Waters, S.M.; Hess, M.; et al. Addressing global ruminant agricultural challenges through understanding the rumen microbiome: Past, present, and future. Front. Microbiol. 2018, 9, 2161. [Google Scholar] [CrossRef]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef]
- Edwards, U.; Rogall, T.; Blocker, H.; Emde, M.; Bottger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Bandarupalli, V.V.K.; St-Pierre, B. Identification of a candidate starch utilizing strain of Prevotella albensis from bovine rumen. Microorganisms 2020, 8, 2005. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Zagnitko, O. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Kumar, S.; Vecchiarelli, B.; Shirley, D.J.; Bittinger, K.; Baker, L.D.; Ferguson, J.D.; Thomsen, N. Temporal dynamics in the ruminal microbiome of dairy cows during the transition period. J. Anim. Sci. 2014, 2, 4014–4022. [Google Scholar] [CrossRef]
- Lima, F.S.; Oikonomou, G.; Lima, S.F.; Bicalho, M.L.S.; Ganda, E.K.; de Oliveira Filho, J.C.; Lorenzo, G.; Trojacanec, P.; Bicalhoa, R.C. Prepartum and postpartum rumen fluid microbiomes: Characterization and correlation with production traits in dairy cows. Appl. Environ. Microbiol. 2015, 81, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Opdahl, L.J.; Gonda, M.G.; St-Pierre, B. Identification of uncultured bacterial species from Firmicutes, Bacteroidetes and CANDIDATUS Saccharibacteria as candidate cellulose utilizers from the rumen of beef cows. Microorganisms 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 2012, 7, e33306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef] [PubMed]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Bryant, M.P.; Small, N. The anaerobic monotrichous butyric acid-producing curved rod-shaped bacteria of the rumen. J. Bacteriol. 1956, 72, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Labutti, K.; Pukall, R.; Steenblock, K.; Glavina Del Rio, T.; Tice, H.; Copeland, A.; Cheng, J.-F.; Lucas, S.; Chen, F.; Nolan, M.; et al. Complete genome sequence of Anaerococcus prevotii type strain (PC1). Stand. Genomic. Sci. 2009, 1, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.B. Fermentation of cellodextrins by cellulolytic and noncellulolytic rumen bacteria. Appl. Environ. Microbiol. 1985, 49, 572–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladd, J.N.; Walker, D.J. The fermentation of lactate and acrylate by the rumen microorganism LC. Biochem. J. 1959, 71, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L.; Wood, W.A.; Emery, R.S. Lactate metabolism by Peptostreptococcus elsdenii: Evidence for lactyl coenzyme a dehydrase. Biochim. Biophys. Acta 1965, 97, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L.; Milligan, L.P. Electron transport in Peptostreptococcus elsdenii. Biochim. Biophys. Acta 1964, 92, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Brockman, H.L.; Wood, W.A. Electron-transferring flavoprotein of Peptostreptococcus elsdenii that functions in the reduction of acrylyl-coenzyme A. J. Bacteriol. 1975, 124, 1447–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counotte, G.H.; Prins, R.A.; Janssen, R.H.; Debie, M.J. Role of Megasphaera elsdenii in the fermentation of dl-[2-13C]lactate in the rumen of dairy cattle. Appl. Environ. Microbiol. 1981, 42, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvie, E.I. Bacterial lactate dehydrogenases. Microbiol. Rev. 1980, 44, 106–139. [Google Scholar] [CrossRef]
- Ricke, S.C.; Martin, S.A.; Nisbet, D.J. Ecology, metabolism, and genetics of ruminal Selenomonads. Crit. Rev. Microbiol. 1996, 22, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, S.; Wang, Y.; Fan, X.; Wang, Y.; Feng, C. Assessment of bacterial community composition and dynamics in alfalfa silages with and without Lactobacillus plantarum inoculation using absolute quantification 16S rRNA sequencing. Front. Microbiol. 2021, 11, 629894. [Google Scholar] [CrossRef]
- Zhang, G.; Fang, X.; Feng, G.; Li, Y.; Zhang, Y. Silage fermentation, bacterial community, and aerobic stability of total mixed ration containing wet corn gluten feed and corn stover prepared with different additives. Animals 2020, 10, 1775. [Google Scholar] [CrossRef]
- Ghali, M.B.; Scott, P.T.; Alhadrami, G.A.; Al Jassim, R.A.M. Identification and characterisation of the predominant lactic acid-producing and lactic acid-utilising bacteria in the foregut of the feral camel (Camelus dromedarius) in Australia. Animm. Prod. Sci. 2011, 51, 597–604. [Google Scholar] [CrossRef]
- Diez-Gonzalez, F.; Bond, D.R.; Jennings, E.; Russell, J.B. Alternative schemes of butyrate production in Butyrivibrio fibrisolvens and their relationship to acetate utilization, lactate production, and phylogeny. Arch. Microbiol. 1999, 171, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.C.; Frick, I.-M. Gram-positive anaerobic cocci--commensals and opportunistic pathogens. FEMS Microbiol. Rev. 2013, 37, 520–553. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.Y.; Oh, B.S.; Ryu, S.W.; Kim, J.-S.; Lee, J.-S.; Park, S.-H.; Kang, S.W.; Lee, J.; Lee, M.-K.; Choe, H.; et al. Anaerococcus faecalis sp. nov., isolated from swine faeces. Curr. Microbiol. 2021, 78, 2589–2594. [Google Scholar] [CrossRef]
- Murdoch, D.A. Gram-positive anaerobic cocci. Clin. Microbiol. Rev. 1998, 11, 81–120. [Google Scholar] [CrossRef] [Green Version]
- Cibis, K.G.; Gneipel, A.; Konig, H. Isolation of acetic, propionic and butyric acid-forming bacteria from biogas plants. J. Biotechnol. 2016, 220, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Dekovic, D.K.; Burschel, S. Assembly of the Escherichia coli NADH:ubiquinone oxidoreductase (respiratory complex I). Biochim. Biophys. Acta 2016, 1857, 214–223. [Google Scholar] [CrossRef]
- Kuhns, M.; Trifunović, D.; Huber, H.; Müller, V. The Rnf complex is a Na+ coupled respiratory enzyme in a fermenting bacterium, Thermotoga maritima. Commun. Biol. 2020, 3, 431. [Google Scholar] [CrossRef]
- Vitt, S.; Prinz, S.; Eisinger, M.; Ermler, U.; Buckel, W. Purification and structural characterization of the Na+-translocating ferredoxin: NAD+ reductase (Rnf) complex of Clostridium tetanomorphum. Nat. Commun. 2022, 13, 6315. [Google Scholar] [CrossRef]
- Ungerfeld, E.M. Metabolic hydrogen flows in rumen fermentation: Principles and possibilities of interventions. Front. Microbiol. 2020, 11, 1–21. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandarupalli, V.V.K.; St-Pierre, B. Metagenomics-Based Analysis of Candidate Lactate Utilizers from the Rumen of Beef Cattle. Microorganisms 2023, 11, 658. https://doi.org/10.3390/microorganisms11030658
Bandarupalli VVK, St-Pierre B. Metagenomics-Based Analysis of Candidate Lactate Utilizers from the Rumen of Beef Cattle. Microorganisms. 2023; 11(3):658. https://doi.org/10.3390/microorganisms11030658
Chicago/Turabian StyleBandarupalli, Venkata Vinay Kumar, and Benoit St-Pierre. 2023. "Metagenomics-Based Analysis of Candidate Lactate Utilizers from the Rumen of Beef Cattle" Microorganisms 11, no. 3: 658. https://doi.org/10.3390/microorganisms11030658
APA StyleBandarupalli, V. V. K., & St-Pierre, B. (2023). Metagenomics-Based Analysis of Candidate Lactate Utilizers from the Rumen of Beef Cattle. Microorganisms, 11(3), 658. https://doi.org/10.3390/microorganisms11030658