
Salinity Impacts the Functional mcrA and dsrA Gene Abundances in Everglades Marshes

Abstract
:1. Introduction
1.1. Saltwater Perturbation
1.2. The Everglades
1.3. Soil Biota and Their Role in Biogeochemical Cycles
1.4. This Study
2. Materials and Methods
2.1. Sample Preparation for Metagenomic Sequencing
2.2. DNA Extraction, NGS Library Construction and Sequencing
2.3. Functional Gene Amplicon Data Processing and Statistical Analysis
2.4. Physiochemical Properties of the Soil Microbiomes
2.5. Statistical Analyses
3. Results
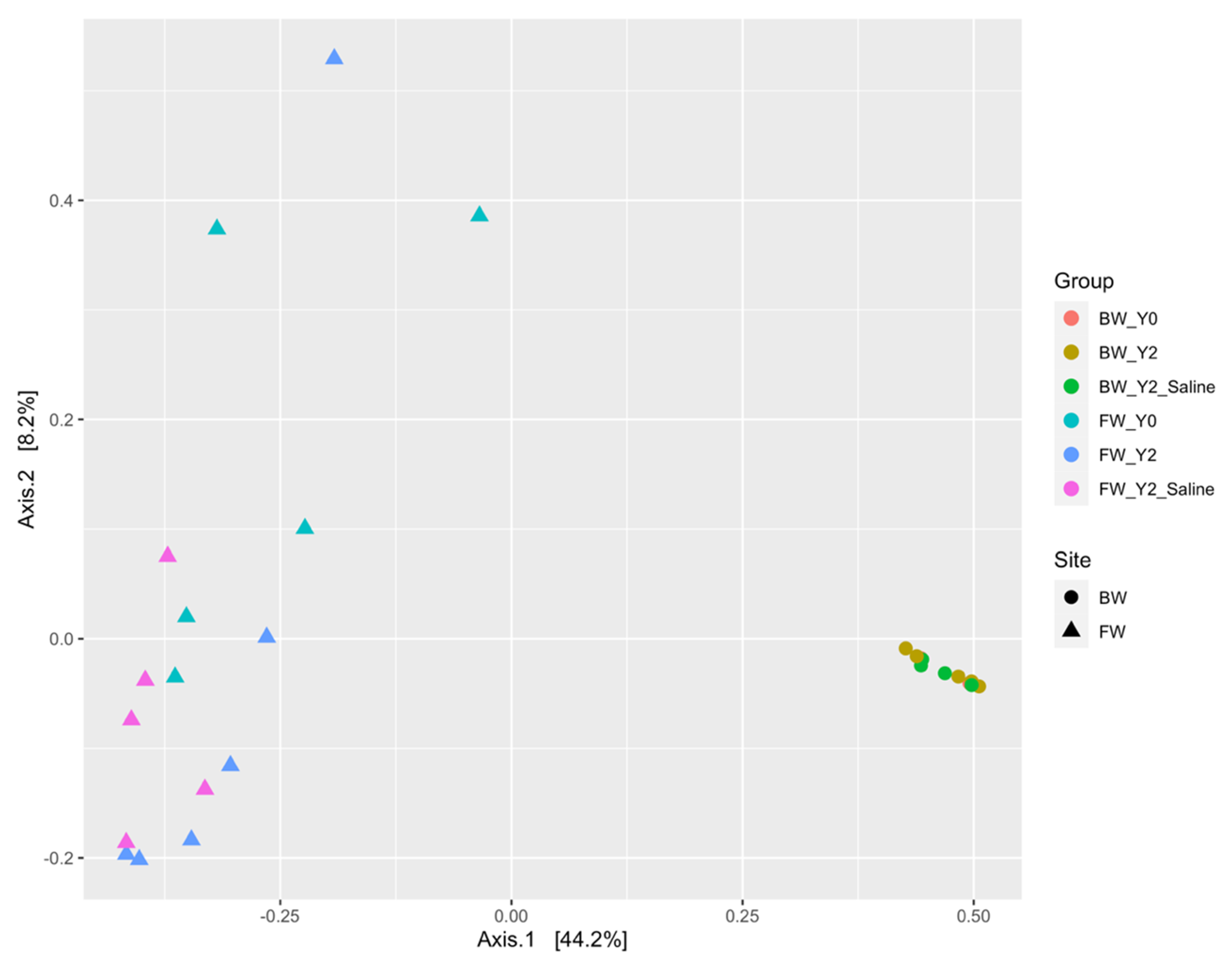
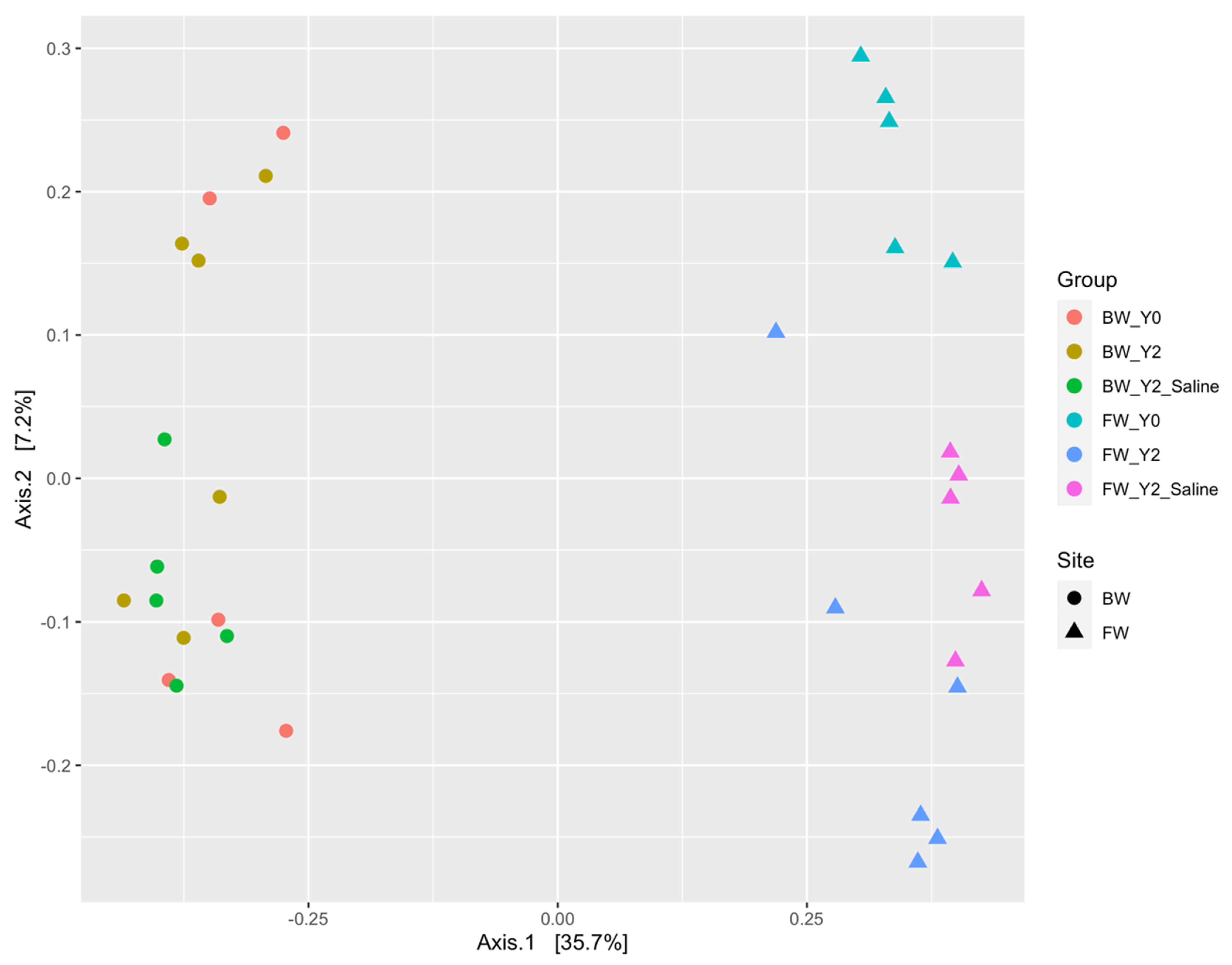
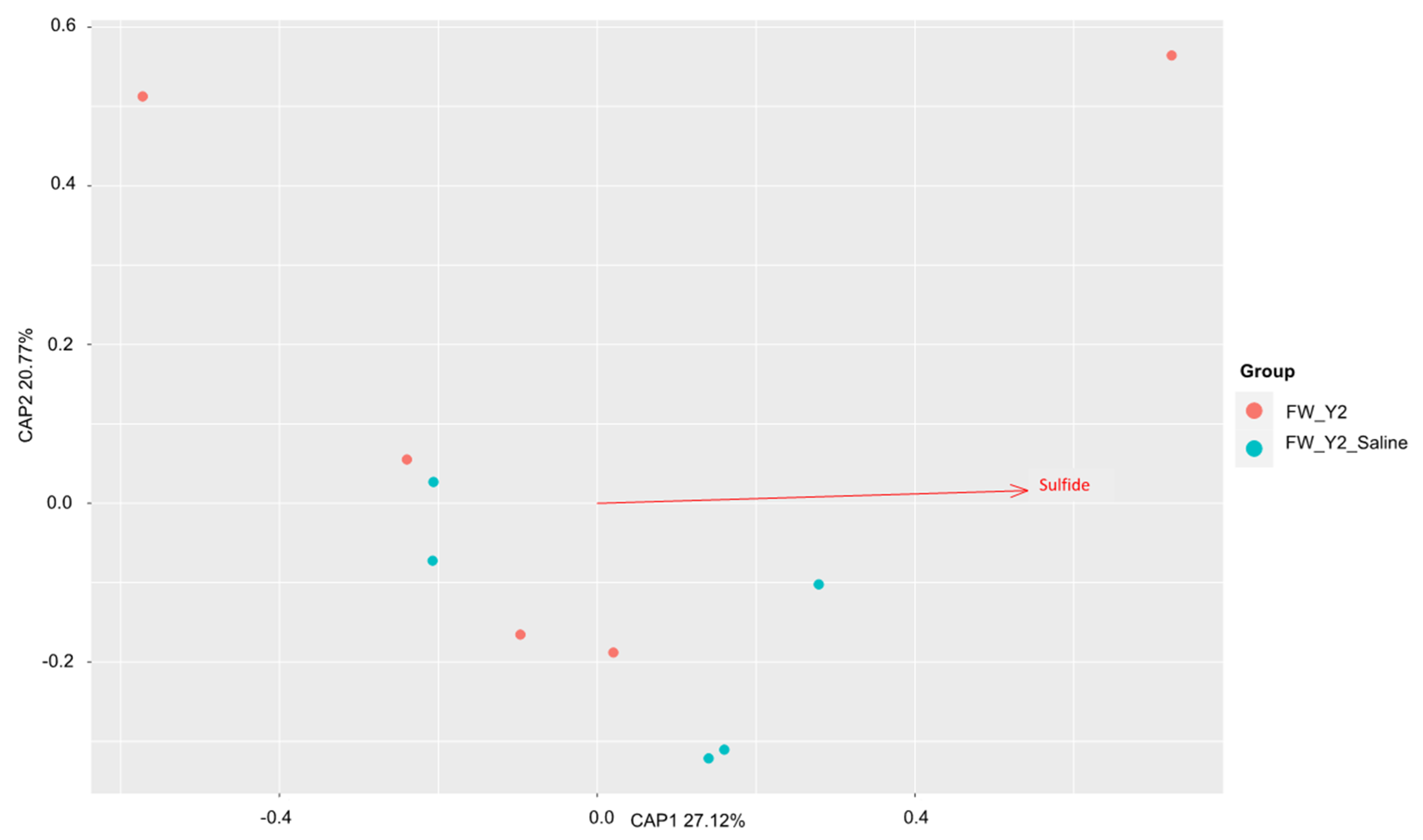
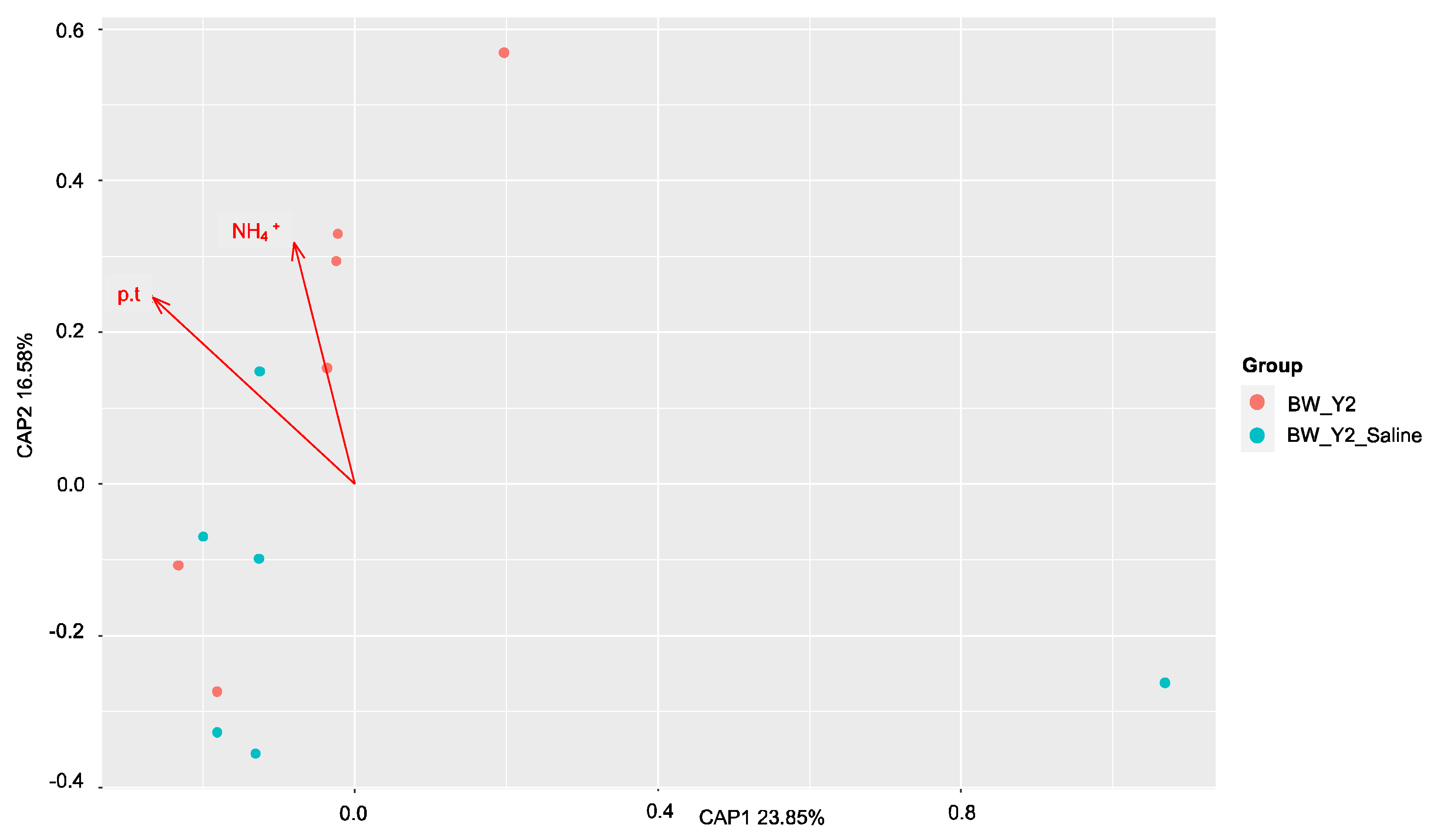
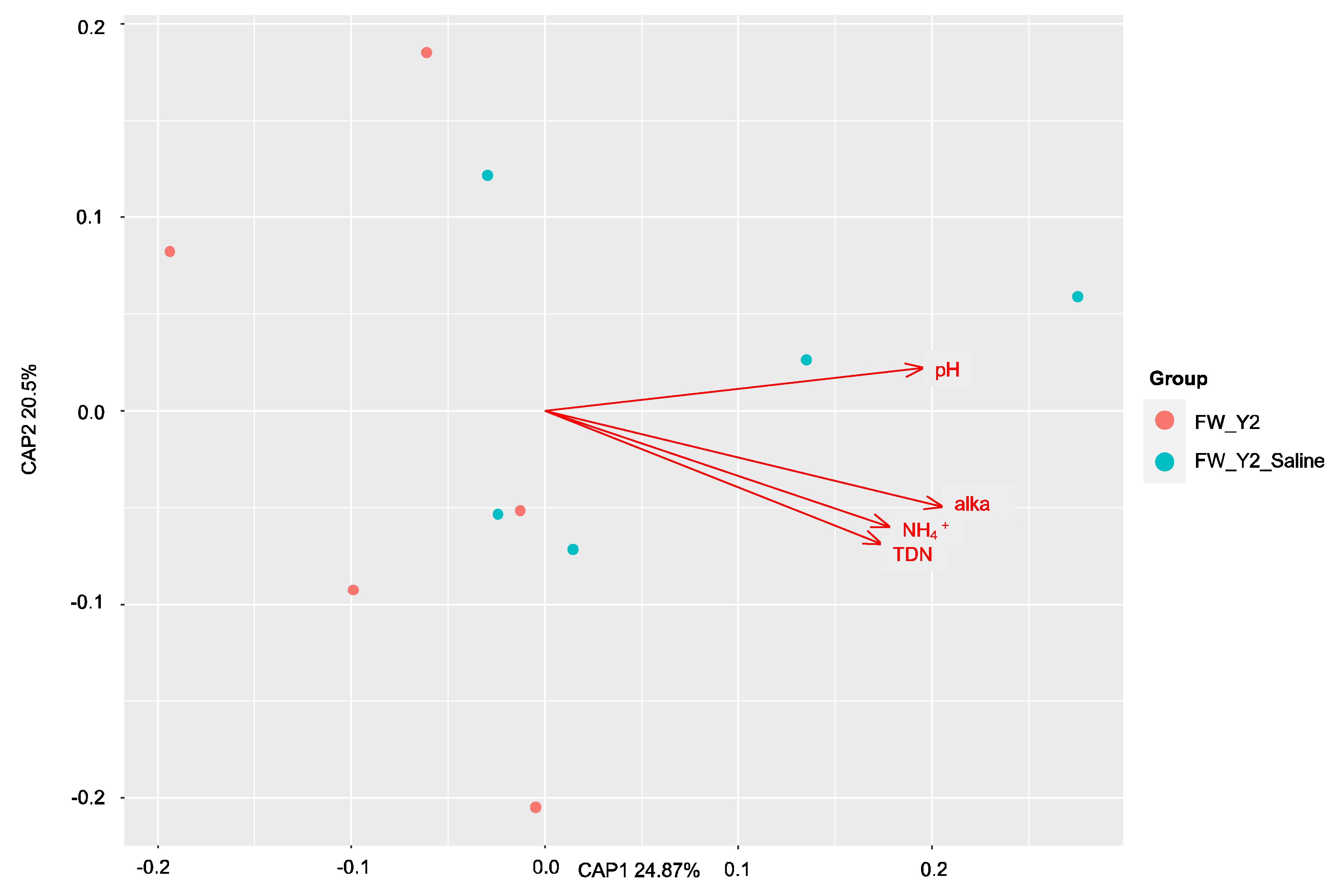
3.1. Beta Diversity Patterns of Functional Genes
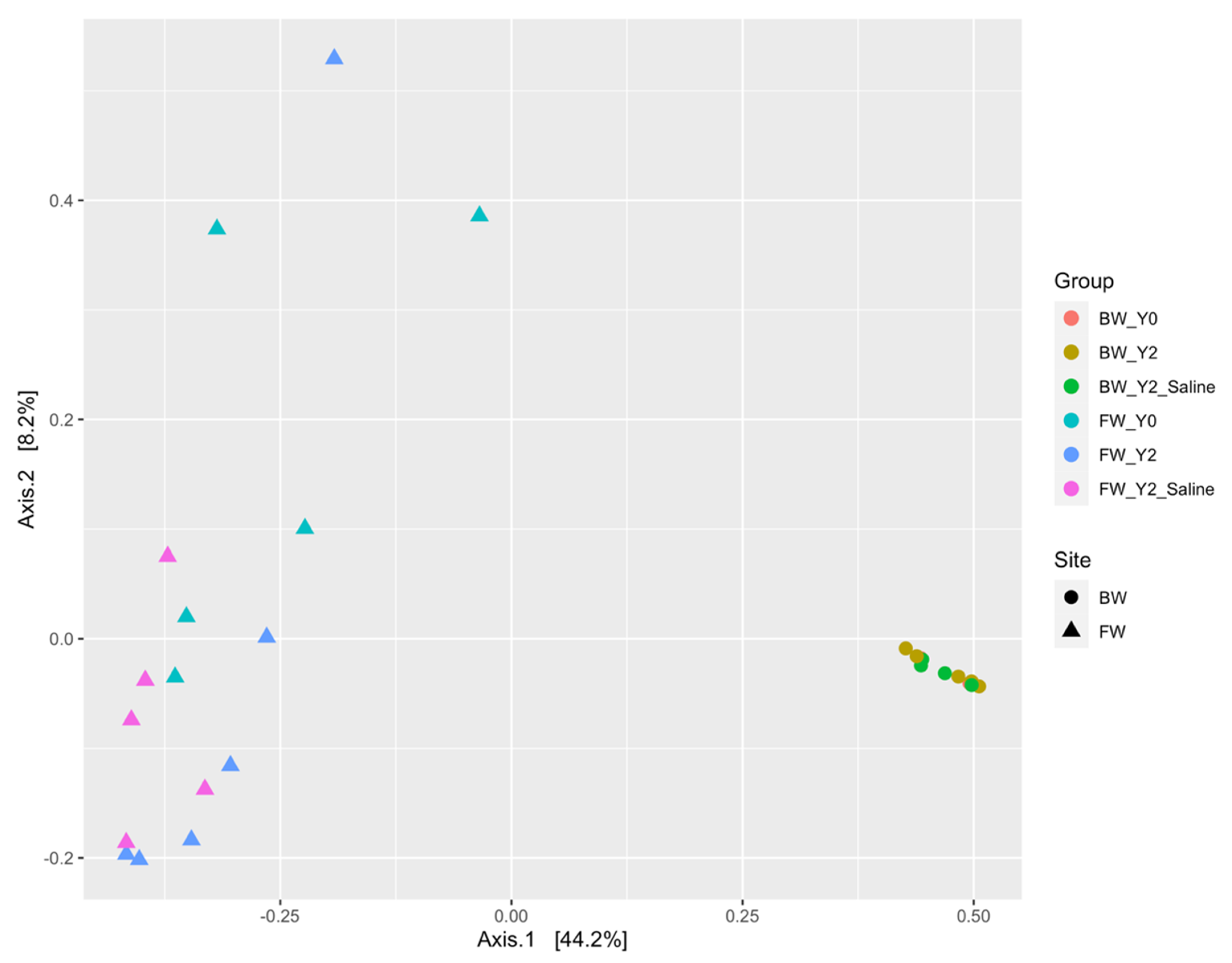
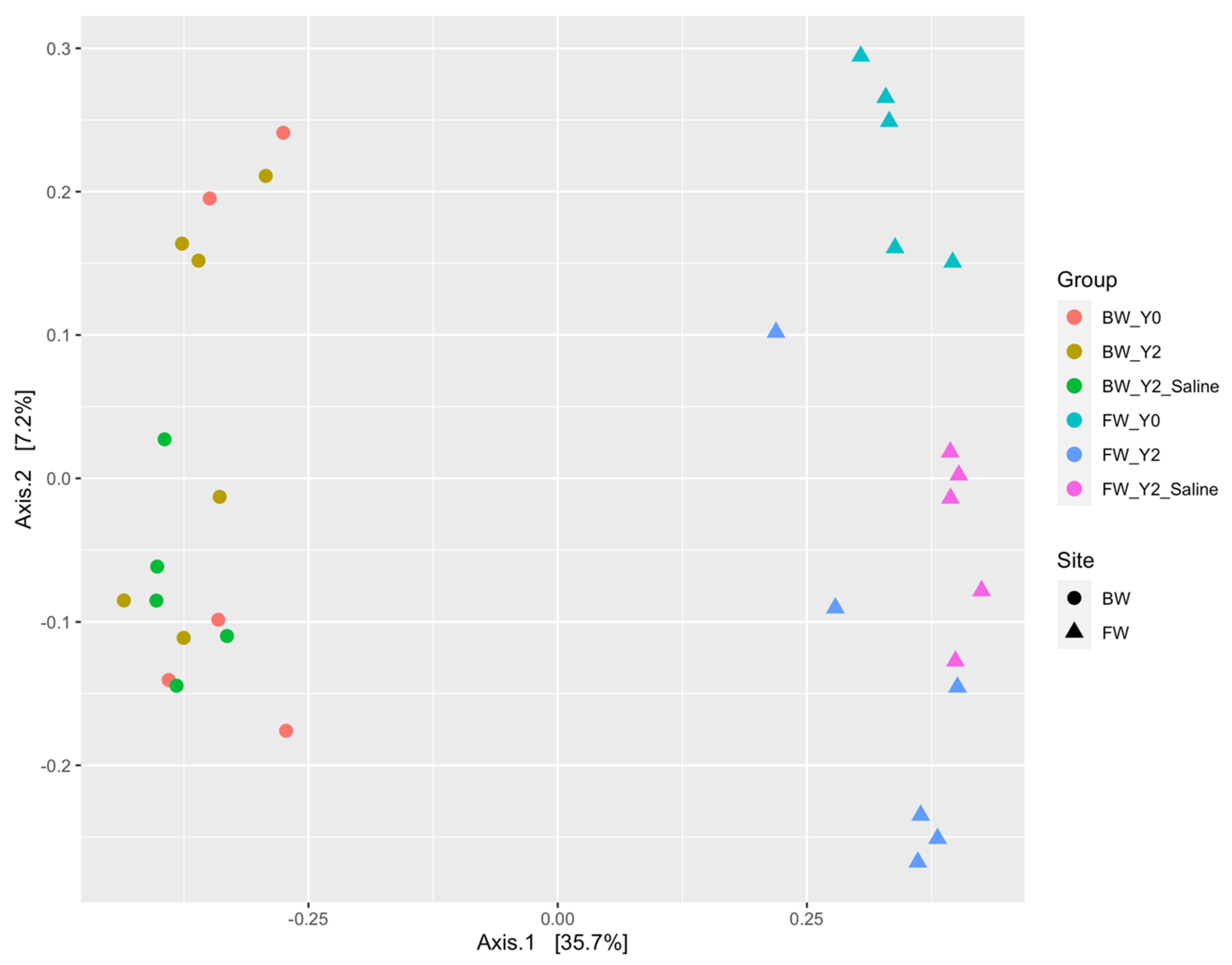
3.1.1. The mcrA Gene
3.1.2. The dsrA Gene
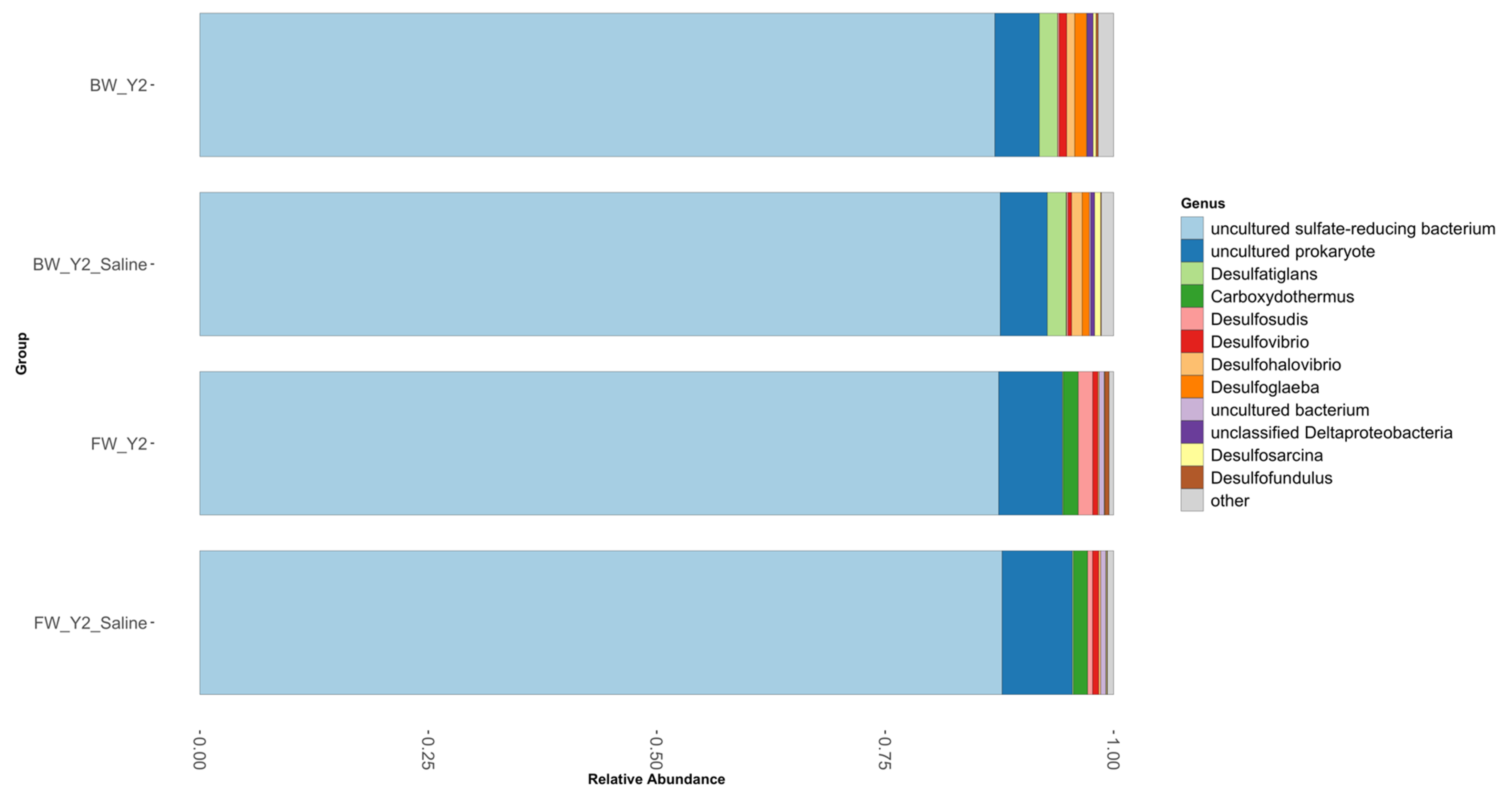
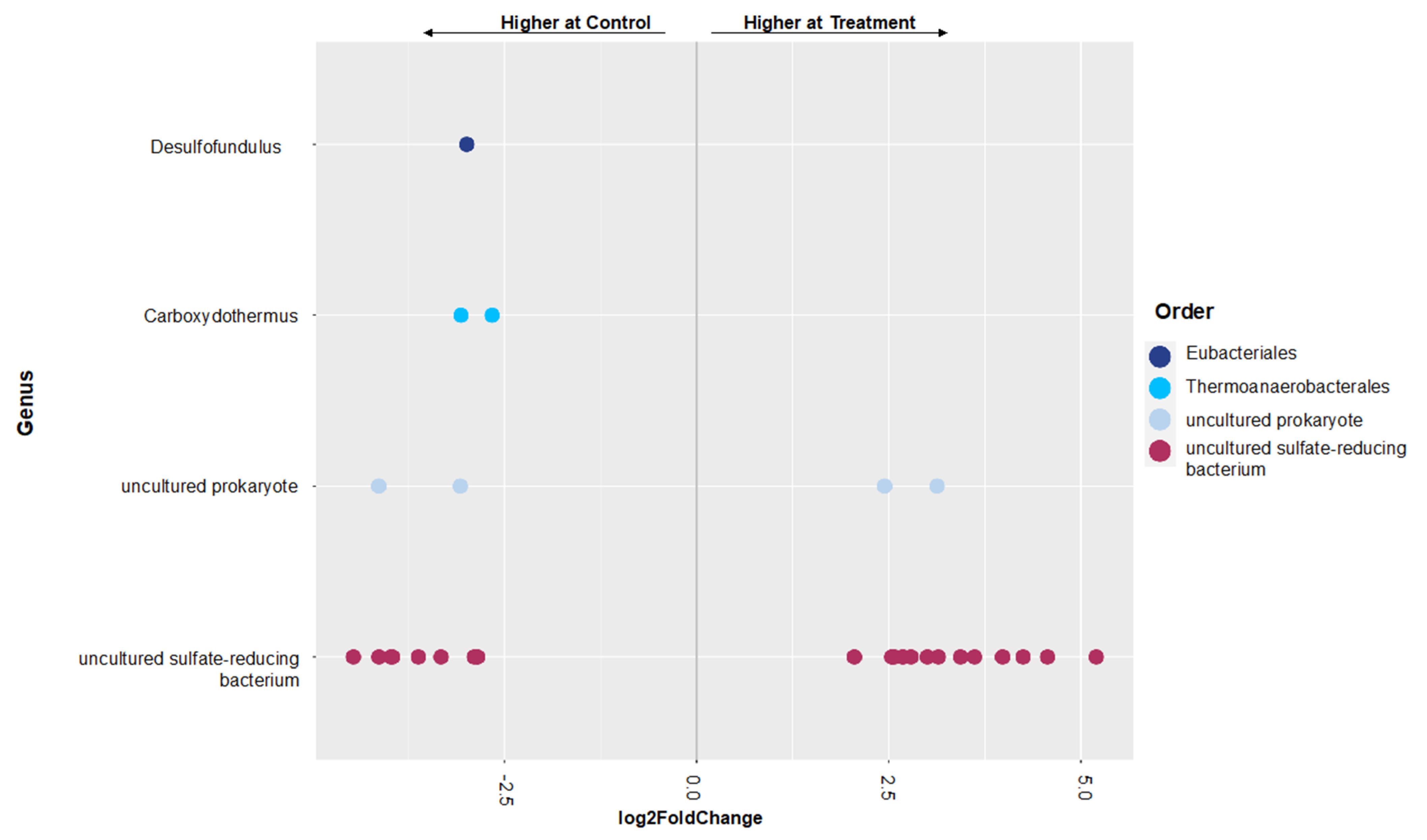
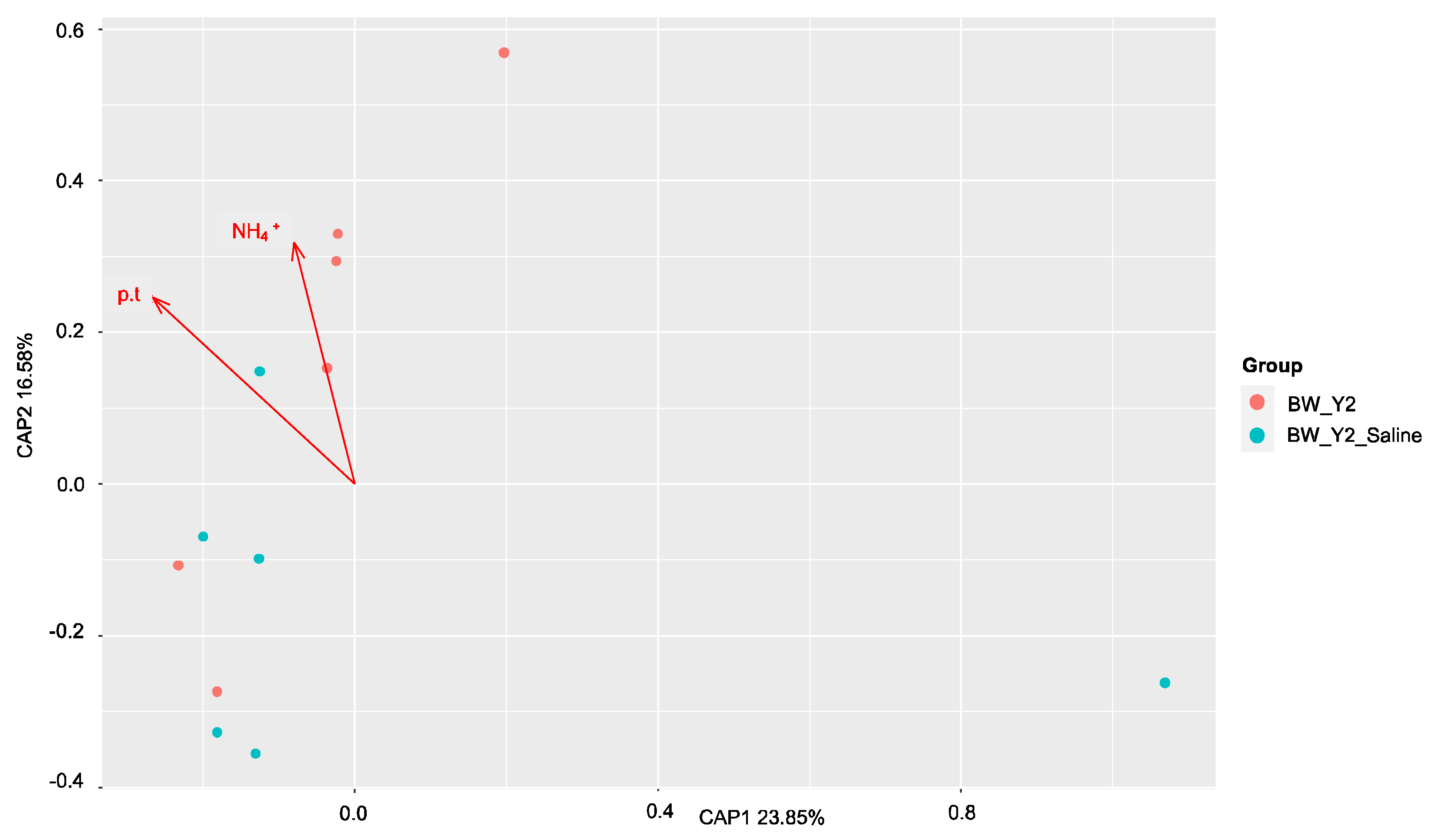
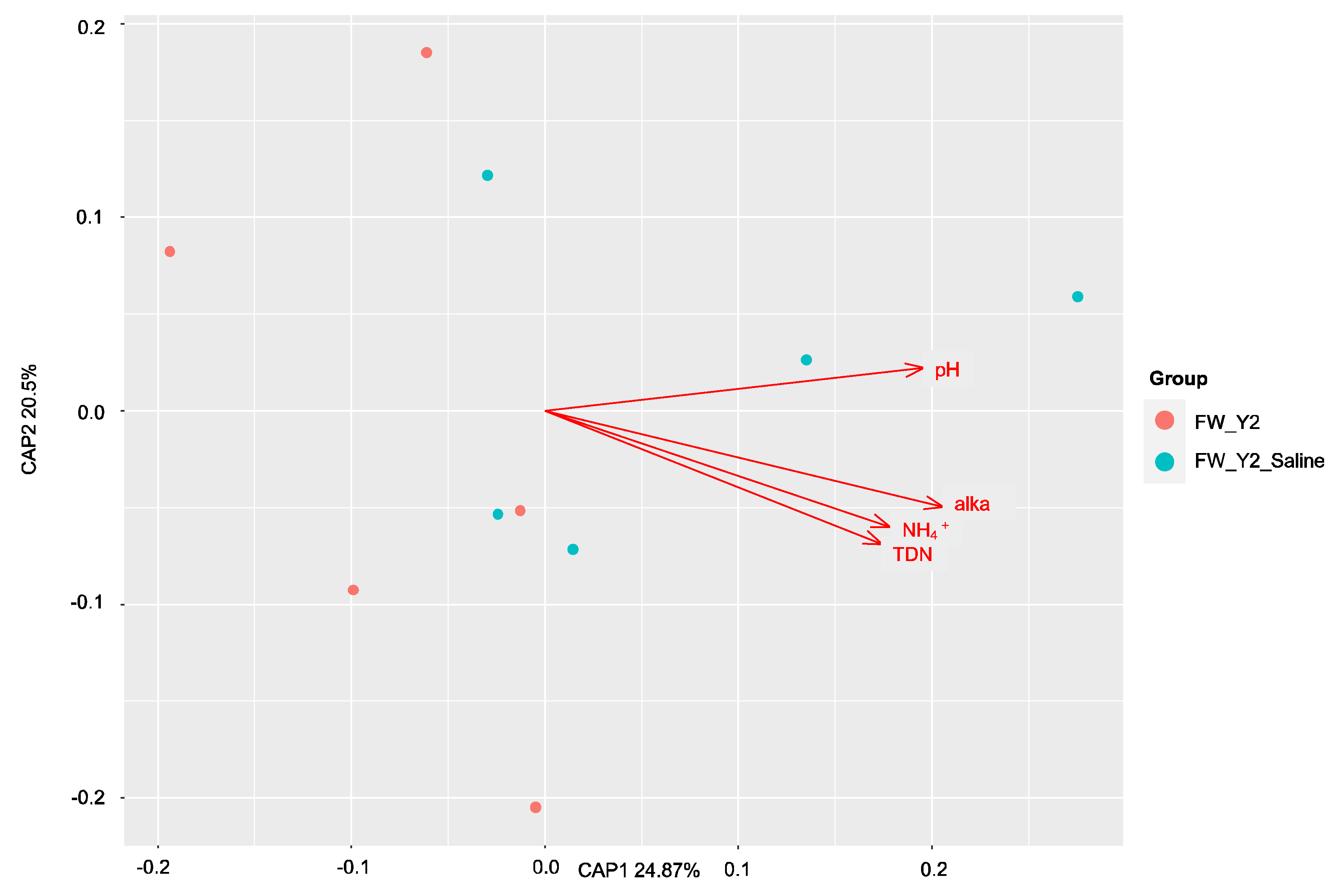
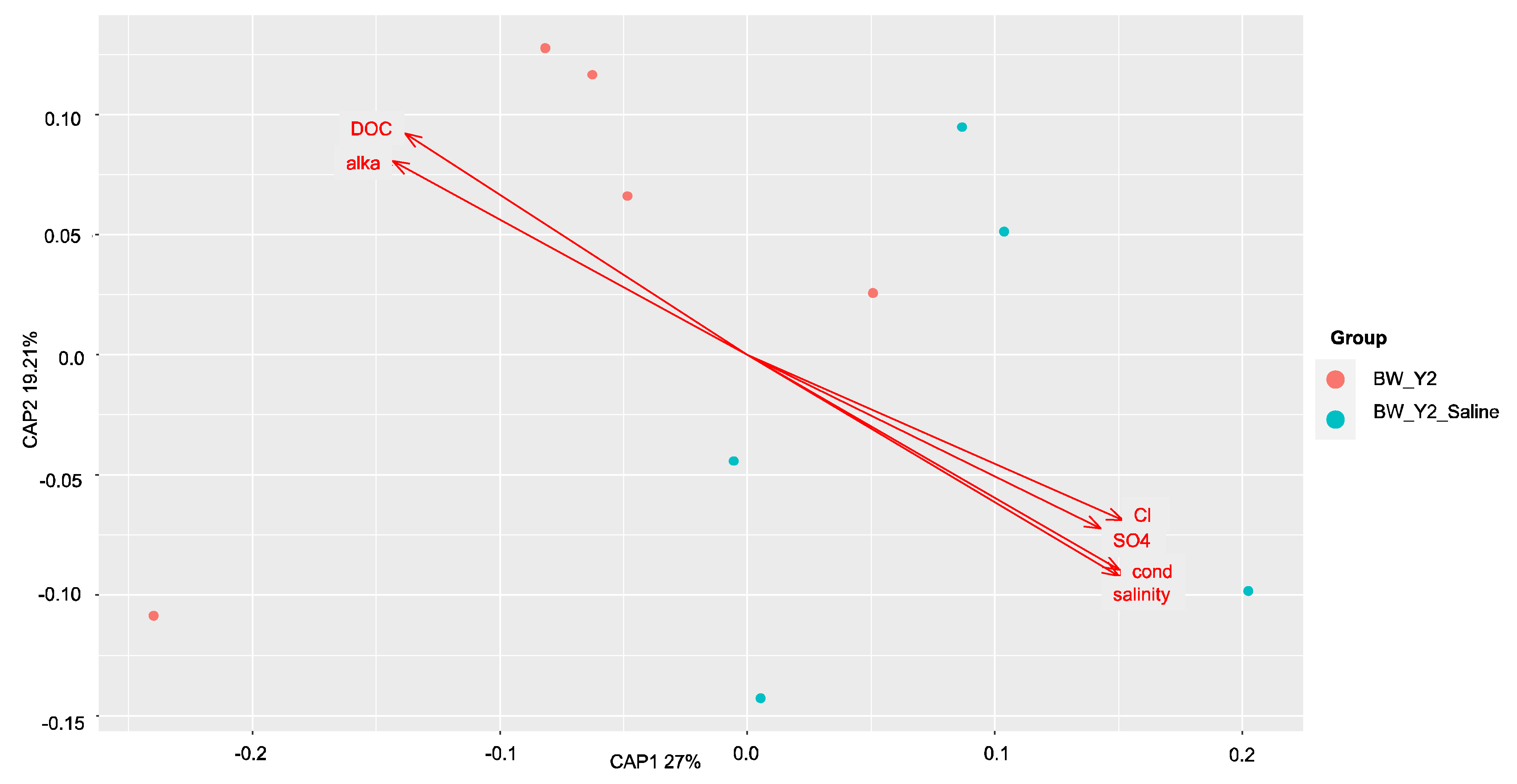
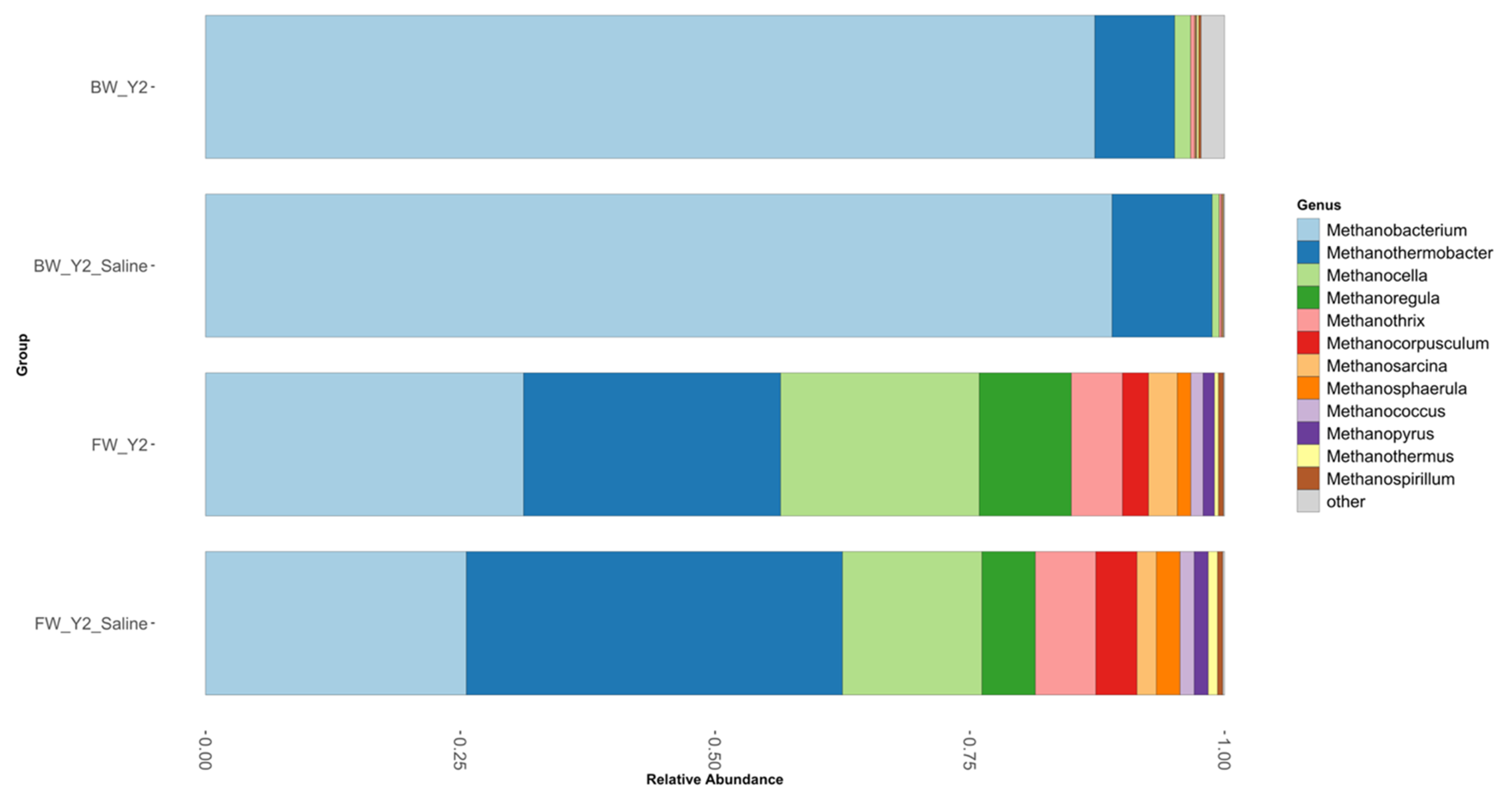
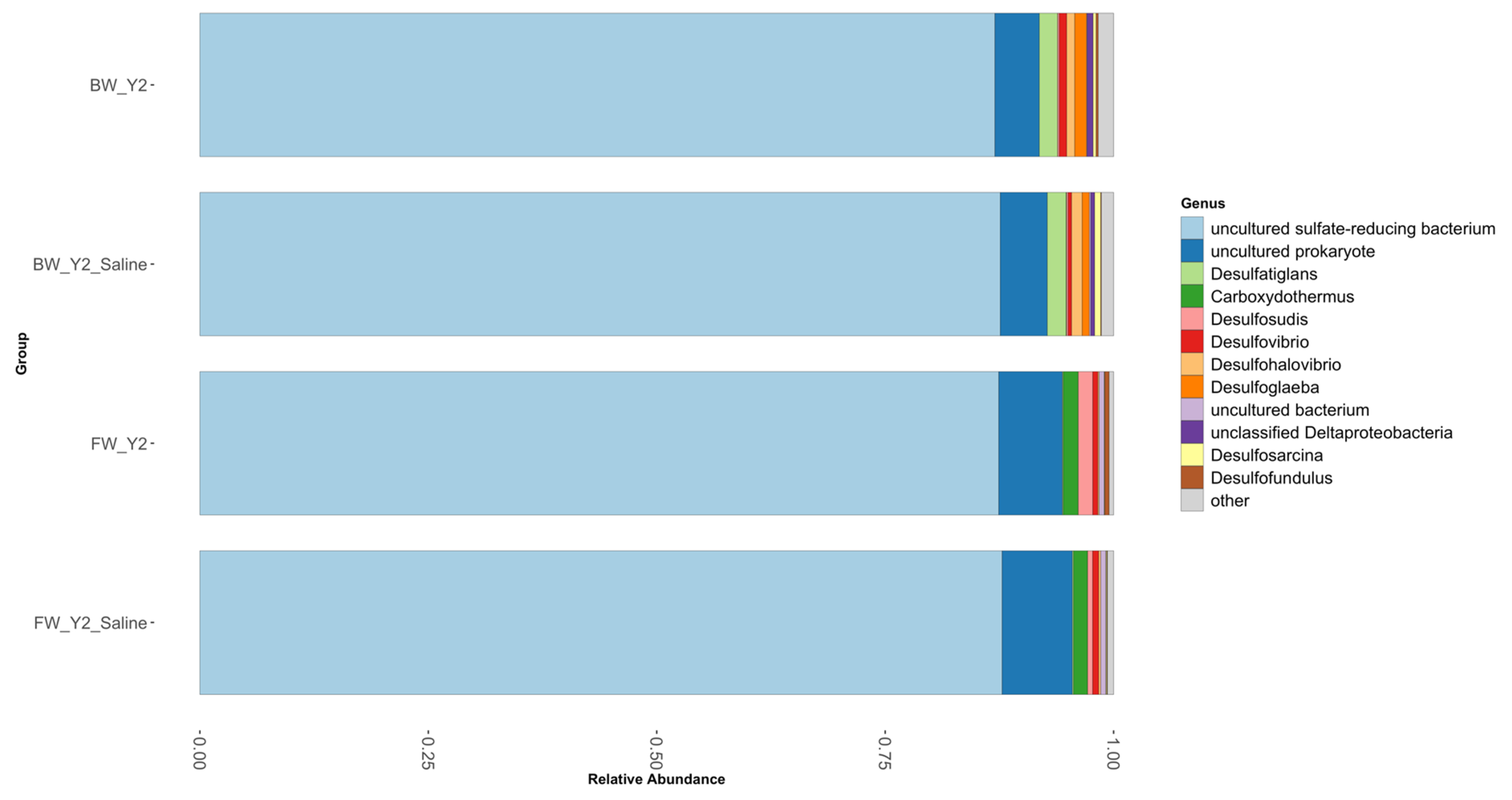
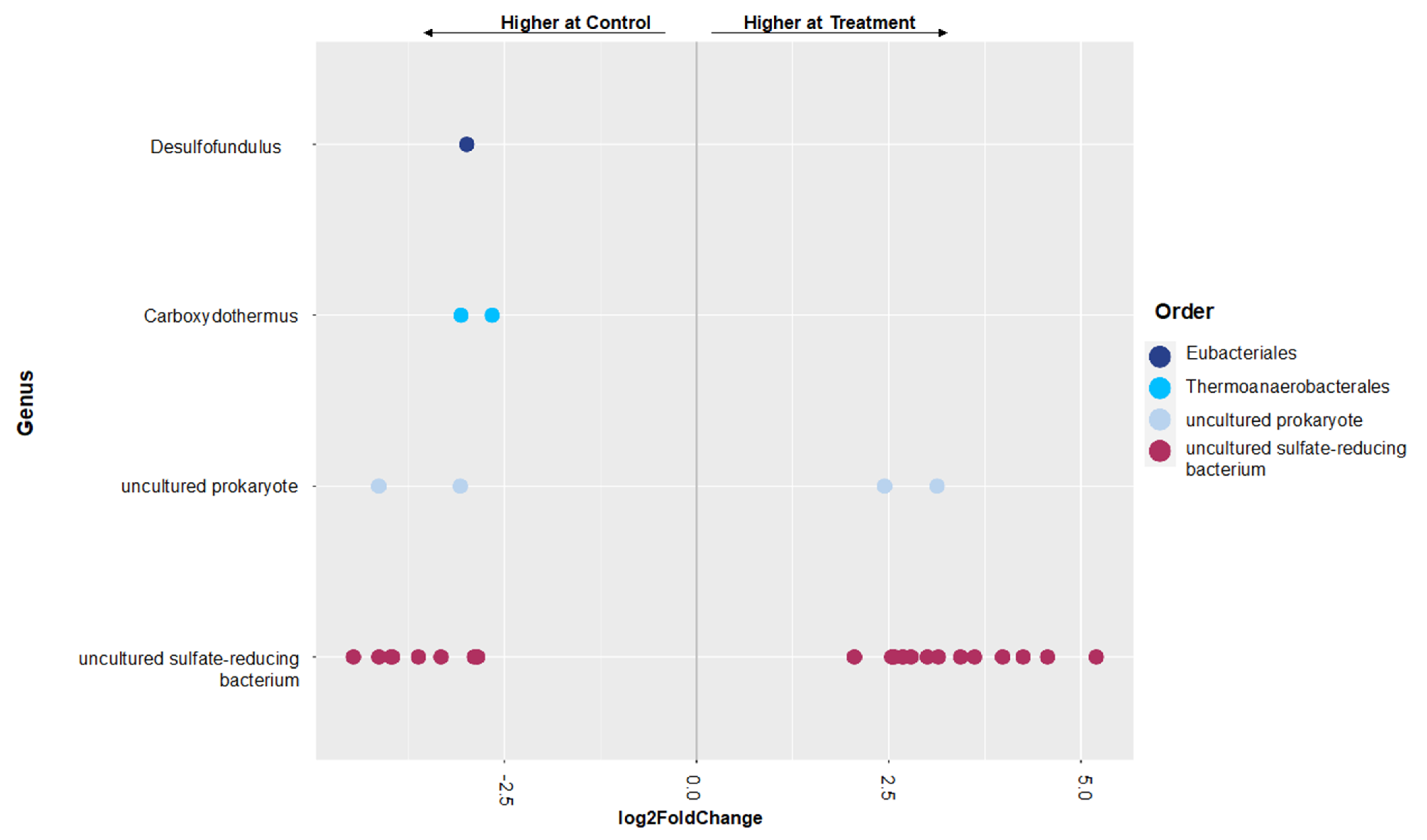
3.2. Shifts in Microbial Functional Potential after Salinization
3.3. Environmental Drivers of the Microbial Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bodelier, P.L.E.; Dedysh, S.N. Microbiology of wetlands. Front. Microbiol. 2013, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Muller, B.; Fuxelius, H.H.; Schnurer, A. AcetoBase: A functional gene repository and database for formyltetrahydrofolate synthetase sequences. Database 2019, 2019, baz142. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.R.; Lubberstedt, T. Functional markers in plants. Trends Plant Sci. 2003, 8, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.K.; Wong, C.K.; Chu, K.H.; Kwan, H.S. Community Structure, Dynamics and Interactions of Bacteria, Archaea and Fungi in Subtropical Coastal Wetland Sediments. Sci. Rep. 2018, 8, 14397. [Google Scholar] [CrossRef]
- Zhang, G.; Bai, J.; Tebbe, C.C.; Zhao, Q.; Jia, J.; Wang, W.; Wang, X.; Yu, L. Salinity controls soil microbial community structure and function in coastal estuarine wetlands. Environ. Microbiol. 2021, 23, 1020–1037. [Google Scholar] [CrossRef]
- Roguet, A.; Eren, A.M.; Newton, R.J.; McLellan, S.L. Fecal source identification using random forest. Microbiome 2018, 6, 185. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Turukalo, T.L.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Ghannam, R.B.; Techtmann, S.M. Machine learning applications in microbial ecology, human microbiome studies, and environmental monitoring. Comput. Struct. Biotechnol. J. 2021, 19, 1092–1107. [Google Scholar] [CrossRef]
- Damaso, N.; Mendel, J.; Mendoza, M.; von Wettberg, E.J.; Narasimhan, G.; Mills, D. Bioinformatics Approach to Assess the Biogeographical Patterns of Soil Communities: The Utility for Soil Provenance. J. Forensic Sci. 2018, 63, 1033–1042. [Google Scholar] [CrossRef]
- Flojgaard, C.; Froslev, T.G.; Brunbjerg, A.K.; Bruun, H.H.; Moeslund, J.; Hansen, A.J.; Ejrnaes, R. Predicting provenance of forensic soil samples: Linking soil to ecological habitats by metabarcoding and supervised classification. PLoS ONE 2019, 14, e0202844. [Google Scholar] [CrossRef]
- Wilhelm, R.C.; van Es, H.M.; Buckley, D.H. Predicting measures of soil health using the microbiome and supervised machine learning. Soil Biol. Biochem. 2022, 164, 108472. [Google Scholar] [CrossRef]
- Chambers, L.G.; Davis, S.E.; Troxler, T.; Boyer, J.N.; Downey-Wall, A.; Scinto, L.J. Biogeochemical effects of simulated sea level rise on carbon loss in an Everglades mangrove peat soil. Hydrobiologia 2013, 726, 195–211. [Google Scholar] [CrossRef]
- Neubauer, S.C.; Piehler, M.F.; Smyth, A.R.; Franklin, R.B. Saltwater Intrusion Modifies Microbial Community Structure and Decreases Denitrification in Tidal Freshwater Marshes. Ecosystems 2018, 22, 912–928. [Google Scholar] [CrossRef]
- Tully, K.; Gedan, K.; Epanchin-Niell, R.; Strong, A.; Bernhardt, E.S.; Bendor, T.; Mitchell, M.; Kominoski, J.; Jordan, T.E.; Neubauer, S.C.; et al. The invisible flood: The chemistry, ecology, and social implications of coastal saltwater intrusion. Bioscience 2019, 69, 368–378. [Google Scholar] [CrossRef]
- Xie, R.; Rao, P.; Pang, Y.; Shi, C.; Li, J.; Shen, D. Salt intrusion alters nitrogen cycling in tidal reaches as determined in field and laboratory investigations. Sci. Total Environ. 2020, 729, 138803. [Google Scholar] [CrossRef]
- Hutchins, D.A.; Jansson, J.K.; Remais, J.V.; Rich, V.I.; Singh, B.K.; Trivedi, P. Climate change microbiology—Problems and perspectives. Nat. Rev. Microbiol. 2019, 17, 391–396. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef]
- Steinman, A.D.; Kindervater, E. Ecosystem restoration in the Everglades and Great Lakes ecosystems: Past, present, and future preventative management. Inland Waters 2020, 12, 8–18. [Google Scholar] [CrossRef]
- Dang, C.; Morrissey, E.M.; Neubauer, S.C.; Franklin, R.B. Novel microbial community composition and carbon biogeochemistry emerge over time following saltwater intrusion in wetlands. Glob. Chang. Biol. 2019, 25, 549–561. [Google Scholar] [CrossRef]
- Galagan, J.E. The Genome of M. acetivorans Reveals Extensive Metabolic and Physiological Diversity. Genome Res. 2002, 12, 532–542. [Google Scholar] [CrossRef]
- Morrissey, E.M.; Gillespie, J.L.; Morina, J.C.; Franklin, R.B. Salinity affects microbial activity and soil organic matter content in tidal wetlands. Glob. Chang. Biol. 2014, 20, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Servais, S.; Kominoski, J.S.; Coronado-Molina, C.; Bauman, L.; Davis, S.E.; Gaiser, E.E.; Kelly, S.; Madden, C.; Mazzei, V.; Rudnik, D.; et al. Effects of saltwater pulses on soil microbial enzymes and organic matter breakdown in freshwater and brackish coastal wetlands. Estuaries Coasts 2020, 43, 814–830. [Google Scholar] [CrossRef]
- Servais, S.; Kominoski, J.S.; Charles, S.P.; Gaiser, E.E.; Mazzei, V.; Troxler, T.G.; Wilson, B.J. Saltwater intrusion and soil carbon loss: Testing effects of salinity and phosphorus loading on microbial functions in experimental freshwater wetlands. Geoderma 2019, 337, 1291–1300. [Google Scholar] [CrossRef]
- Offre, P.; Spang, A.; Schleper, C. Archaea in Biogeochemical Cycles. Annu. Rev. Microbiol. 2013, 67, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, E.M.; Franklin, R.B. Evolutionary history influences the salinity preference of bacterial taxa in wetland soils. Front. Microbiol. 2015, 6, 1013. [Google Scholar] [CrossRef]
- Chambers, L.G.; Davis, S.E.; Troxler, T.G. Microbiology of the Everglades Ecosystem; James, A., Entry, A.D.G., Krish, J., Andrew, O., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 89–112. [Google Scholar]
- Bingman, M.J.; Atkinson, C. Elemental composition of commercial seasalts. J. Aquaricult. Aquat. Sci. 1997, 8, 39–43. [Google Scholar]
- Morris, R.; Schauer-Gimenez, A.; Bhattad, U.; Kearney, C.; Struble, C.A.; Zitomer, D.; Maki, J.S. Methyl coenzyme M reductase (mcrA) gene abundance correlates with activity measurements of methanogenic H2/CO2-enriched anaerobic biomass. Microb. Biotechnol. 2014, 7, 77–84. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for Illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef]
- Fish, J.A.; Chai, B.; Wang, Q.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. FunGene: The functional gene pipeline and repository. Front. Microbiol. 2013, 4, 291. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Wang, Q.; Quensen, J.F., 3rd; Fish, J.A.; Lee, T.K.; Sun, Y.; Tiedje, J.M.; Cole, J.R. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio 2013, 4, e00592-13. [Google Scholar] [CrossRef]
- R Core Team. R: A language and environment for statistical computing. R Found. Stat. Comput. 2021, 12, 12. [Google Scholar]
- Quensen, J. RDPutils: R Utilities for processing RDPTool output. R Package Version 2018, 1. Available online: https://rdrr.io/github/jfq3/RDPutils/ (accessed on 1 April 2023).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Jupke, J.F.; Schafer, R.B. Should ecologists prefer model- over distance-based multivariate methods? Ecol. Evol. 2020, 10, 2417–2435. [Google Scholar] [CrossRef]
- Legendre, P.; Fortin, M.J. Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Mol. Ecol. Resour. 2010, 10, 831–844. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; Wagner, H.; et al. Vegan: Community Ecology Package. 2020. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 1 April 2023).
- Robert, C.K.; Somerfield, P.J.; Airoldi, L.; Warwick, R.M. Exploring interactions by second-stage community analyses. J. Exp. Mar. Biol. Ecol. 2006, 338, 179–192. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Sela-Adler, M.; Ronen, Z.; Herut, B.; Antler, G.; Vigderovich, H.; Eckert, W.; Sivan, O. Co-existence of Methanogenesis and Sulfate Reduction with Common Substrates in Sulfate-Rich Estuarine Sediments. Front. Microbiol. 2017, 8, 766. [Google Scholar] [CrossRef]
- Gao, M.; Yang, J.; Liu, Y.; Zhang, J.; Li, J.; Liu, Y.; Wu, B.; Gu, L. Deep insights into the anaerobic co-digestion of waste activated sludge with concentrated leachate under different salinity stresses. Sci. Total Environ. 2022, 838, 155922. [Google Scholar] [CrossRef]
- Louca, S.; Polz, M.F.; Mazel, F.; Albright, M.B.; Huber, J.A.; O’Connor, M.I.; Ackermann, M.; Hahn, A.S.; Srivastava, D.S.; Crowe, S.A.; et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2018, 2, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Nold, S.C.; Zwart, G. Patterns and governing forces in aquatic microbial communities. Aquat. Ecol. 1998, 32, 17–35. [Google Scholar] [CrossRef]
- Fei, X.X.; Wang, L.; Jun, H.J.; Shu, T.Y.; Hu, Y.; Hua, F.X.; Sun, Y.; Fai, T.Y.; Nan, Z.Y.; Hai, C.J. Salinity influence on soil microbial respiration rate of wetland in the Yangtze River estuary through changing microbial community. J. Environ. Sci. 2014, 26, 2562–2570. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L.; Jiang, H.; Wu, G.; Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016, 6, 25078. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, I.A.; Grasby, S.E.; Haupt, E.S.; Dunfield, P.F. Analysis of microbial communities in natural halite springs reveals a domain-dependent relationship of species diversity to osmotic stress. Environ. Microbiol. Rep. 2018, 10, 695–703. [Google Scholar] [CrossRef]
- Liu, Y.; Whitman, W.B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 2008, 1125, 171–189. [Google Scholar] [CrossRef]
- Oremland, R.S.; Polcin, S. Methanogenesis and sulfate reduction: Competitive and noncompetitive substrates in estuarine sediments. Appl. Environ. Microbiol. 1982, 44, 1270–1276. [Google Scholar] [CrossRef]
- Liu, D.; Ding, W.; Jia, Z.; Cai, Z. The impact of dissolved organic carbon on the spatial variability of methanogenic archaea communities in natural wetland ecosystems across China. Appl. Microbiol. Biotechnol. 2012, 96, 253–263. [Google Scholar] [CrossRef]
- Zhao, J.; Chakrabarti, S.; Chambers, R.; Weisenhorn, P.; Travieso, R.; Stumpf, S.; Standen, E.; Briceno, H.; Troxler, T.; Gaiser, E.; et al. Year-around survey and manipulation experiments reveal differential sensitivities of soil prokaryotic and fungal communities to saltwater intrusion in Florida Everglades wetlands. Sci. Total Environ. 2023, 858, 159865. [Google Scholar] [CrossRef]
- An, J.; Liu, C.; Wang, Q.; Yao, M.; Rui, J.; Zhang, S.; Li, X. Soil bacterial community structure in Chinese wetlands. Geoderma 2019, 337, 290–299. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Gawas, V.S.; Shivaramu, M.S.; Damare, S.R.; Pujitha, D.; Meena, R.M.; Shenoy, B.D. Diversity and extracellular enzyme activities of heterotrophic bacteria from sediments of the Central Indian Ocean Basin. Sci. Rep. 2019, 9, 9403. [Google Scholar] [CrossRef]
- Tong, C.; Cadillo-Quiroz, H.; Zeng, Z.H.; She, C.X.; Yang, P.; Huang, J.F. Changes of community structure and abundance of methanogens in soils along a freshwater–brackish water gradient in subtropical estuarine marshes. Geoderma 2017, 299, 101–110. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temporal | Treatment | |||
|---|---|---|---|---|
| BW_Y0 vs. BW_Y2 | FW_Y0 vs. FW_Y2 | BW_Y2 vs. BW_Y2_Saline | FW_Y2 vs. FW_Y2_Saline | |
| Gene | ||||
| mcrA | R = 0.291, p > 0.05 | R = 0.151, p > 0.05 | R = 0.319, p = 0.051 | R = 0.093, p > 0.05 |
| dsrA | R = 0.307, p < 0.05 | R = 0.608, p < 0.05 | R = -0.045, p > 0.05 | R = 0.227, p < 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, D.; Kominoski, J.S.; Servais, S.; Mills, D. Salinity Impacts the Functional mcrA and dsrA Gene Abundances in Everglades Marshes. Microorganisms 2023, 11, 1180. https://doi.org/10.3390/microorganisms11051180
Jordan D, Kominoski JS, Servais S, Mills D. Salinity Impacts the Functional mcrA and dsrA Gene Abundances in Everglades Marshes. Microorganisms. 2023; 11(5):1180. https://doi.org/10.3390/microorganisms11051180
Chicago/Turabian StyleJordan, Deidra, John S. Kominoski, Shelby Servais, and DeEtta Mills. 2023. "Salinity Impacts the Functional mcrA and dsrA Gene Abundances in Everglades Marshes" Microorganisms 11, no. 5: 1180. https://doi.org/10.3390/microorganisms11051180
APA StyleJordan, D., Kominoski, J. S., Servais, S., & Mills, D. (2023). Salinity Impacts the Functional mcrA and dsrA Gene Abundances in Everglades Marshes. Microorganisms, 11(5), 1180. https://doi.org/10.3390/microorganisms11051180