
In Silico and In Vitro Investigation of the Distribution and Expression of Key Genes in the Fucose Operon of Escherichia coli


Abstract
1. Introduction
2. Materials and Methods
2.1. Microorganisms
2.2. Phenotypic Detection of Fucose Utilization
2.2.1. Carbohydrate Degradation by E. coli Wild-Type and Mutant Strains
2.2.2. Molecular Identification of Fucose Utilization Genes by the Polymerase Chain Reaction
2.2.3. Nucleotide Sequence of Amplified Fucose Utilization Genes
2.3. Subsystems Annotation Using SEED Database
2.4. Comparative Genomic Analysis
2.5. In Silico Gene Expression Analysis
2.6. RNA Extraction
2.7. Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR)
2.8. Statistical Analysis
3. Results
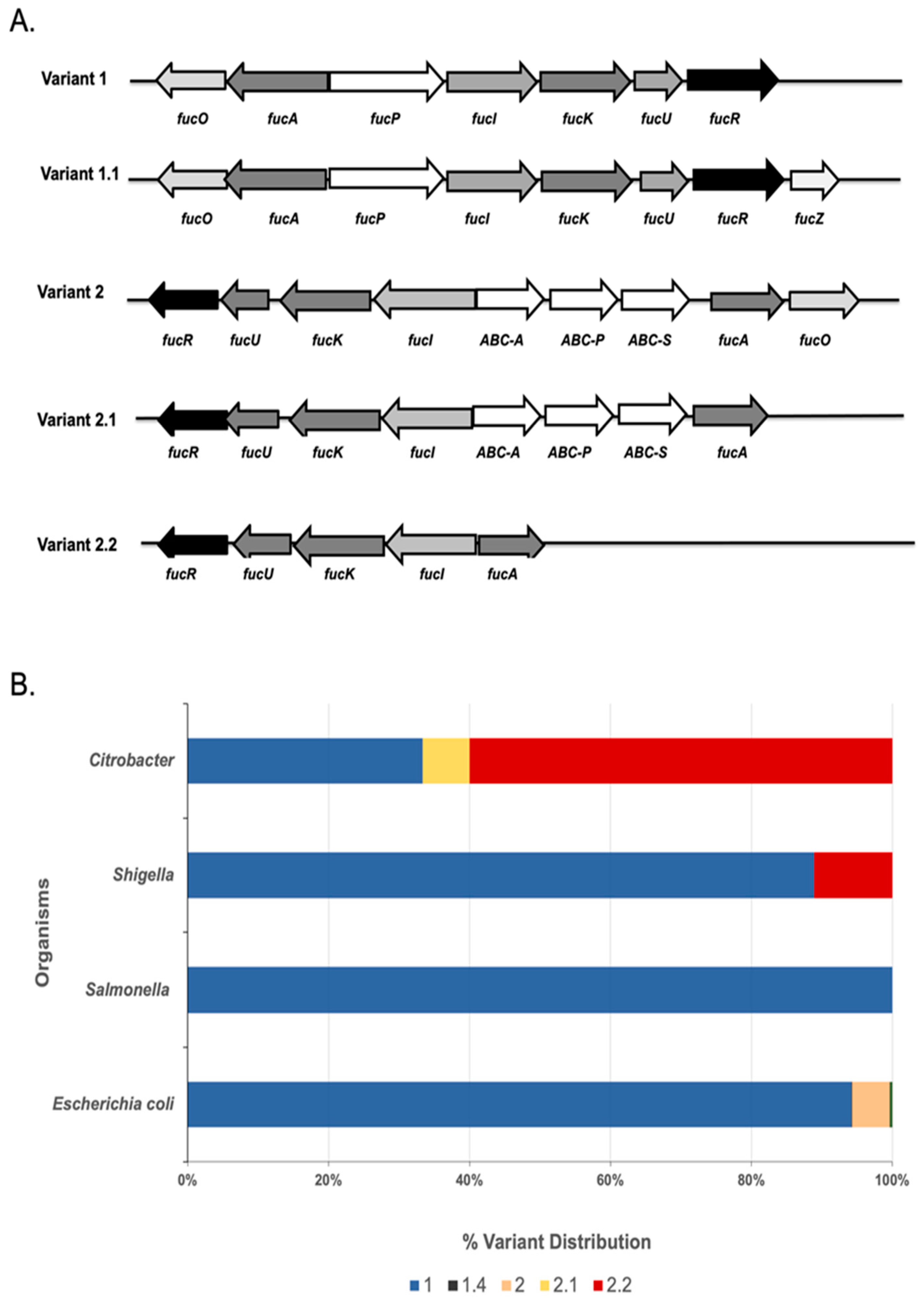
3.1. Comparative Genomics and Subsystems Analysis Uncover Pathway Variants of Fucose Utilization among Different Members of Enterobacteriaceae
3.2. In Vitro Screening of Genetic Determinants Involved in Fucose Sugar Utilization
3.3. Differential Utilization of Glucose and Fucose by Wild-Type and Mutant E. coli Strains
3.4. In Silico Meta-Analysis of Gene Expression Data Reveals the High Expression of fucP in Selected Sugars and Growth Phase
3.5. In Vitro Expression Analysis of Fucose Utilization Genes in M9 Minimal Medium Supplemented with Glucose and Fucose
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pacheco, A.R.; Curtis, M.M.; Ritchie, J.M.; Munera, D.; Waldor, M.K.; Moreira, C.G.; Sperandio, V. Fucose sensing regulates bacterial intestinal colonization. Nature 2012, 492, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 2015, 18, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Staib, L.; Fuchs, T.M. From food to cell: Nutrient exploitation strategies of enteropathogens. Microbiology 2014, 160, 1020–1039. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Ventura, M.; Buttó, L.F.; Duranti, S.; O’Toole, P.W.; Motherway, M.O.C.; van Sinderen, D. Molecular dialogue between the human gut microbiota and the host: A Lactobacillus and Bifidobacterium perspective. Cell. Mol. Life Sci. 2014, 71, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Suzuki, H.; Herlekar, D.; Sartor, R.B.; Campbell, B.J.; Roberts, C.L.; Stewart, K.; Scherl, E.J.; Araz, Y.; Bitar, P.P.; et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm. Bowel Dis. 2014, 20, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Conway, T.; Cohen, P.S. Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiol. Spectr. 2015, 3, 3. [Google Scholar] [CrossRef]
- Petit, E.; LaTouf, W.G.; Coppi, M.V.; Warnick, T.A.; Currie, D.; Romashko, I.; Deshpande, S.; Haas, K.; Alvelo-Maurosa, J.G.; Wardman, C.; et al. Involvement of a bacterial microcompartment in the metabolism of fucose and rhamnose by Clostridium phytofermentans. PLoS ONE 2013, 8, e54337. [Google Scholar] [CrossRef]
- Seemann, J.E.; Schulz, G.E. Structure and mechanism of L-fucose isomerase from Escherichia coli. J. Mol. Biol. 1997, 273, 256–268. [Google Scholar] [CrossRef]
- Aziz, R.K.; Monk, J.M.; Andrews, K.A.; Nhan, J.; Khaw, V.L.; Wong, H.; Palsson, B.O.; Charusanti, P. The aldehyde dehydrogenase, AldA, is essential for L-1,2-propanediol utilization in laboratory-evolved Escherichia coli. Microbiol. Res. 2017, 194, 47–52. [Google Scholar] [CrossRef]
- Aziz, R.K.; Khaw, V.L.; Monk, J.M.; Brunk, E.; Lewis, R.; Loh, S.I.; Mishra, A.; Nagle, A.A.; Satyanarayana, C.; Dhakshinamoorthy, S.; et al. Model-driven discovery of synergistic inhibitors against E. coli and S. enterica serovar Typhimurium targeting a novel synthetic lethal pair, aldA and prpC. Front. Microbiol. 2015, 6, 958. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.M.; Eisenreich, W.; Heesemann, J.; Goebel, W. Metabolic adaptation of human pathogenic and related nonpathogenic bacteria to extra- and intracellular habitats. FEMS Microbiol. Rev. 2012, 36, 435–462. [Google Scholar] [CrossRef] [PubMed]
- Staib, L.; Fuchs, T.M. Regulation of fucose and 1,2-propanediol utilization by Salmonella enterica serovar Typhimurium. Front. Microbiol. 2015, 6, 1116. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Marisch, K.; Bayer, K.; Scharl, T.; Mairhofer, J.; Krempl, P.M.; Hummel, K.; Razzazi-Fazeli, E.; Striedner, G. A comparative analysis of industrial Escherichia coli K–12 and B strains in high-glucose batch cultivations on process-, transcriptome- and proteome level. PLoS ONE 2013, 8, e70516. [Google Scholar] [CrossRef]
- Fong, N.L.; Lerman, J.A.; Lam, I.; Palsson, B.O.; Charusanti, P. Reconciling a Salmonella enterica metabolic model with experimental data confirms that overexpression of the glyoxylate shunt can rescue a lethal ppc deletion mutant. FEMS Microbiol. Lett. 2013, 342, 62–69. [Google Scholar] [CrossRef]
- Muraoka, W.T.; Zhang, Q. Phenotypic and genotypic evidence for L-fucose utilization by Campylobacter jejuni. J. Bacteriol. 2011, 193, 1065–1075. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Yang, C.; Li, X.; Rodionova, I.A.; Wang, Y.; Obraztsova, A.Y.; Zagnitko, O.P.; Overbeek, R.; Romine, M.F.; Reed, S.; et al. Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genom. 2010, 11, 494. [Google Scholar] [CrossRef]
- Aziz, R.K.; Devoid, S.; Disz, T.; Edwards, R.A.; Henry, C.S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. SEED servers: High-performance access to the SEED genomes, annotations, and metabolic models. PLoS ONE 2012, 7, e48053. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Martorell-Marugán, J.; López-Domínguez, R.; García-Moreno, A.; González-Rumayor, V.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019, 35, 880–882. [Google Scholar] [CrossRef]
- Racine, J.S. RStudio: A Platform-Independent IDE for R and Sweave. J. Appl. Econom. 2012, 27, 167–172. [Google Scholar] [CrossRef]
- Zhou, K.; Zhou, L.; Lim, Q.; Zou, R.; Stephanopoulos, G.; Too, H.P. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol. Biol. 2011, 12, 18. [Google Scholar] [CrossRef]
- Lee, D.H.; Palsson, B.O. Adaptive evolution of Escherichia coli K-12 MG1655 during growth on a nonnative carbon source, L-1,2-propanediol. Appl. Environ. Microbiol. 2010, 76, 4158–4168. [Google Scholar] [CrossRef] [PubMed]
- Kittana, H.; Gomes-Neto, J.C.; Heck, K.; Geis, A.L.; Segura Muñoz, R.R.; Cody, L.A.; Schmaltz, R.J.; Bindels, L.B.; Sinha, R.; Hostetter, J.M.; et al. Commensal Escherichia coli strains can promote intestinal inflammation via differential interleukin-6 production. Front. Immunol. 2018, 9, 2318. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef] [PubMed]
- ElRakaiby, M.; Dutilh, B.E.; Rizkallah, M.R.; Boleij, A.; Cole, J.N.; Aziz, R.K. Pharmacomicrobiomics: The impact of human microbiome variations on systems pharmacology and personalized therapeutics. Omics A J. Integr. Biol. 2014, 18, 402–414. [Google Scholar] [CrossRef]
- Pagliari, D.; Piccirillo, C.A.; Larbi, A.; Cianci, R. The interactions between innate immunity and microbiota in gastrointestinal diseases. J. Immunol. Res. 2015, 2015, 898297. [Google Scholar] [CrossRef]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Bin, P.; Tang, Z.; Liu, S.; Chen, S.; Xia, Y.; Liu, J.; Wu, H.; Zhu, G. Intestinal microbiota mediates enterotoxigenic Escherichia coli-induced diarrhea in piglets. BMC Vet. Res. 2018, 14, 385. [Google Scholar] [CrossRef]
- Iversen, H.; Lindbäck, T.; L’Abée-Lund, T.M.; Roos, N.; Aspholm, M.; Stenfors Arnesen, L. The gut bacterium Bacteroides thetaiotaomicron influences the virulence potential of the enterohemorrhagic Escherichia coli O103:H25. PLoS ONE 2015, 10, e0118140. [Google Scholar] [CrossRef]
- Ramos, S.; Silva, V.; Dapkevicius, M.L.E.; Caniça, M.; Tejedor-Junco, M.T.; Igrejas, G.; Poeta, P. Escherichia coli as commensal and pathogenic bacteria among food-producing animals: Health implications of extended spectrum β-lactamase (ESBL) production. Animals 2020, 10, 2239. [Google Scholar] [CrossRef]
- Becerra, J.E.; Yebra, M.J.; Monedero, V. An L-fucose operon in the probiotic Lactobacillus rhamnosus GG Is involved in adaptation to gastrointestinal conditions. Appl. Environ. Microbiol. 2015, 81, 3880–3888. [Google Scholar] [CrossRef]
- Yoon, S.H.; Han, M.J.; Jeong, H.; Lee, C.H.; Xia, X.X.; Lee, D.H.; Shim, J.H.; Lee, S.Y.; Oh, T.K.; Kim, J.F. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012, 13, R37. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, E.A.; Mortlock, R.P. Molecular cloning of the Escherichia coli B L-fucose-D-arabinose gene cluster. J. Bacteriol. 1994, 176, 7223–7232. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheong, Y.E.; Jung, I.; Kim, K.H. Metabolomic and transcriptomic analyses of Escherichia coli for efficient fermentation of L-fucose. Mar. Drugs 2019, 17, 82. [Google Scholar] [CrossRef]
- Park, D.; Choi, D.; Kwak, J.; Park, C.; Ryu, K.-S. Characterization and role of fucose mutarotase in mammalian cells. Glycobiology 2007, 17, 955–962. [Google Scholar] [CrossRef]
- Vital, M.; Chai, B.; Østman, B.; Cole, J.; Konstantinidis, K.T.; Tiedje, J.M. Gene expression analysis of E. coli strains provides insights into the role of gene regulation in diversification. ISME J. 2015, 9, 1130–1140. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Target Gene | Primer Sequence (5′-3′) | Use | Product Length |
|---|---|---|---|---|
| fucP_F | fucP | TTCTGAAACGGGCATGAAAT | PCR | 1697 |
| fucP_R | CCGTAGCTTTCGCCATATTC | |||
| fucI_F | fucI | ATTATTGGCGGCGGTATTG | PCR | 2097 |
| fucI_R | CACAGTCGAGTACCAGGATAA | |||
| eco1_F | eco1 | CGCGAGGAAWATRCCGATAA | PCR | 427 |
| eco1_R | CCTGACHGATAAYGCCTTTCT | |||
| yjfF _f | yjfF | GAGCTCAGCGTRCCATCAAA | PCR | 831 |
| yjfF _R | CGTCTTTGTGYTGGGYTATCT | |||
| fucIB-F | fucI_B | CCACTGGACCGATCAATATC | PCR | 444 |
| fucIB_R | AGGAAGCGGGAAGAGTAA | |||
| fucP_F | fucP_RT | TYATTGGCGGCGGTATTG | RT- PCR | 124 |
| fucP_R | CGGAAACGGGCAAAGATAA | |||
| fucI_F | fucI_RT | CGCCAGCATGAAAGGTAA | RT- PCR | 137 |
| fucI_R | GACGCAGTTCGGTCATATC | |||
| ihfB_F | IhfB | GATAGAAAGACTTGCCACCCA | RT- PCR | 200 |
| ihfB_R | CCAGTTCTACTTTATCGCCAG |
| Variant | Description |
|---|---|
| 1 | The entire operon is present (fucP, fucI, fucK, fucU, fucR, fucA, fucO) |
| 1.1 | The Fuc operon is present + fucZ |
| 1.2 | A variant of 1 that lacks fucO but has fucZ |
| 1.3 | Fucose operon lacking fucU |
| 1.4 | Fucose operon lacking fucO → no anaerobic production of 1, 2-PDO |
| 2 | A variant of the fucose operon that lacks fucP, and has an ABC transporter-encoding set of genes, in most cases |
| 2.1 | A variant of 2 missing fucO |
| 2.2 | Fucose operon missing fucP without a defined ABC transporter permease |
| 3 | Fuc operon + predicted phosphotransferase EIIA and EIIB |
| 4 | Fucase + Fuc operon without fucU + RR1 |
| 4.1 | Fucase + Fuc operon lacking fucP and no RR1 |
| 4.2 | Fucase + Fuc operon but lacking fucO |
| 4.3 | Fucase + Fuc operon |
| 5 | Missing fucR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saif, N.A.; Hashem, Y.A.; Amin, H.M.; Aziz, R.K. In Silico and In Vitro Investigation of the Distribution and Expression of Key Genes in the Fucose Operon of Escherichia coli. Microorganisms 2023, 11, 1265. https://doi.org/10.3390/microorganisms11051265
Saif NA, Hashem YA, Amin HM, Aziz RK. In Silico and In Vitro Investigation of the Distribution and Expression of Key Genes in the Fucose Operon of Escherichia coli. Microorganisms. 2023; 11(5):1265. https://doi.org/10.3390/microorganisms11051265
Chicago/Turabian StyleSaif, Nehal A., Yomna A. Hashem, Heba M. Amin, and Ramy K. Aziz. 2023. "In Silico and In Vitro Investigation of the Distribution and Expression of Key Genes in the Fucose Operon of Escherichia coli" Microorganisms 11, no. 5: 1265. https://doi.org/10.3390/microorganisms11051265
APA StyleSaif, N. A., Hashem, Y. A., Amin, H. M., & Aziz, R. K. (2023). In Silico and In Vitro Investigation of the Distribution and Expression of Key Genes in the Fucose Operon of Escherichia coli. Microorganisms, 11(5), 1265. https://doi.org/10.3390/microorganisms11051265