
A Bittersweet Kiss of Gram-Negative Bacteria: The Role of ADP-Heptose in the Pathogenesis of Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. LPS and Mechanisms of Its Recognition by the Immune System
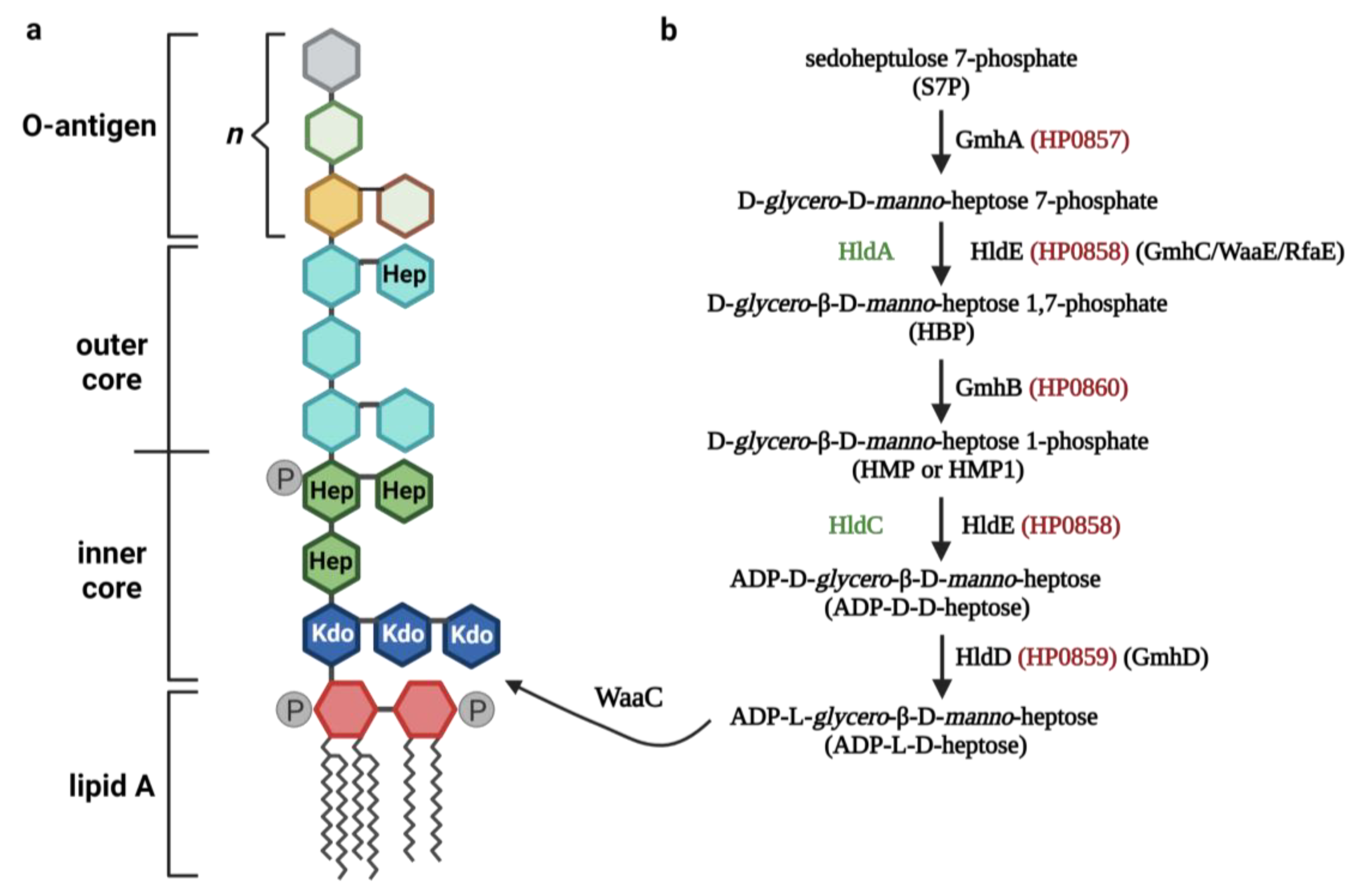
3. ADP-Heptose Metabolism
4. The Road from Recognition of the Role of HMP and HBP to the Discovery of ADP-Heptose as a Novel PAMP
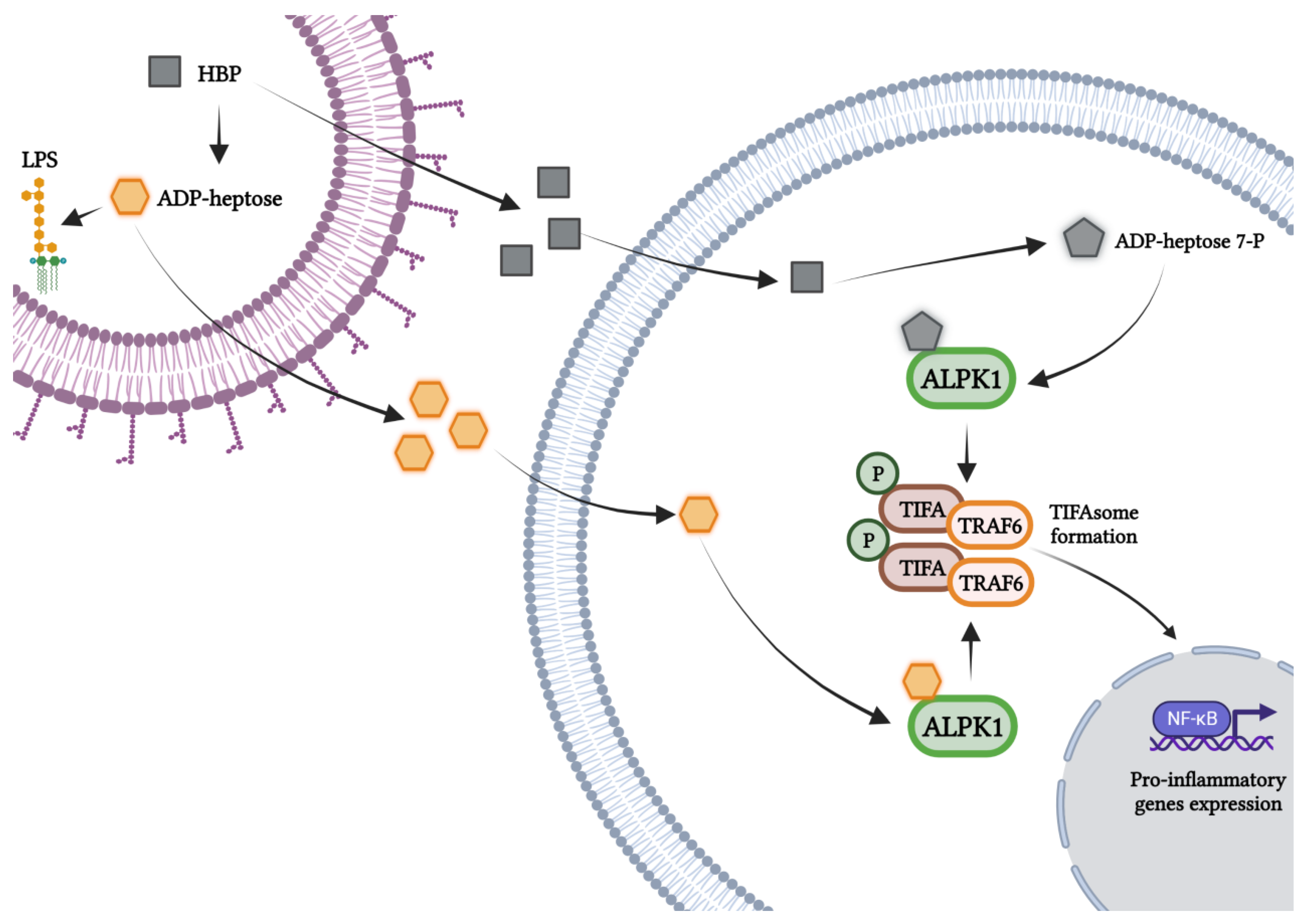
5. Mechanisms of Activation of the ALPK1/TIFA/NF-κB Axis by ADP-Heptose
6. Biological Effects of ADP-Heptose Recognition
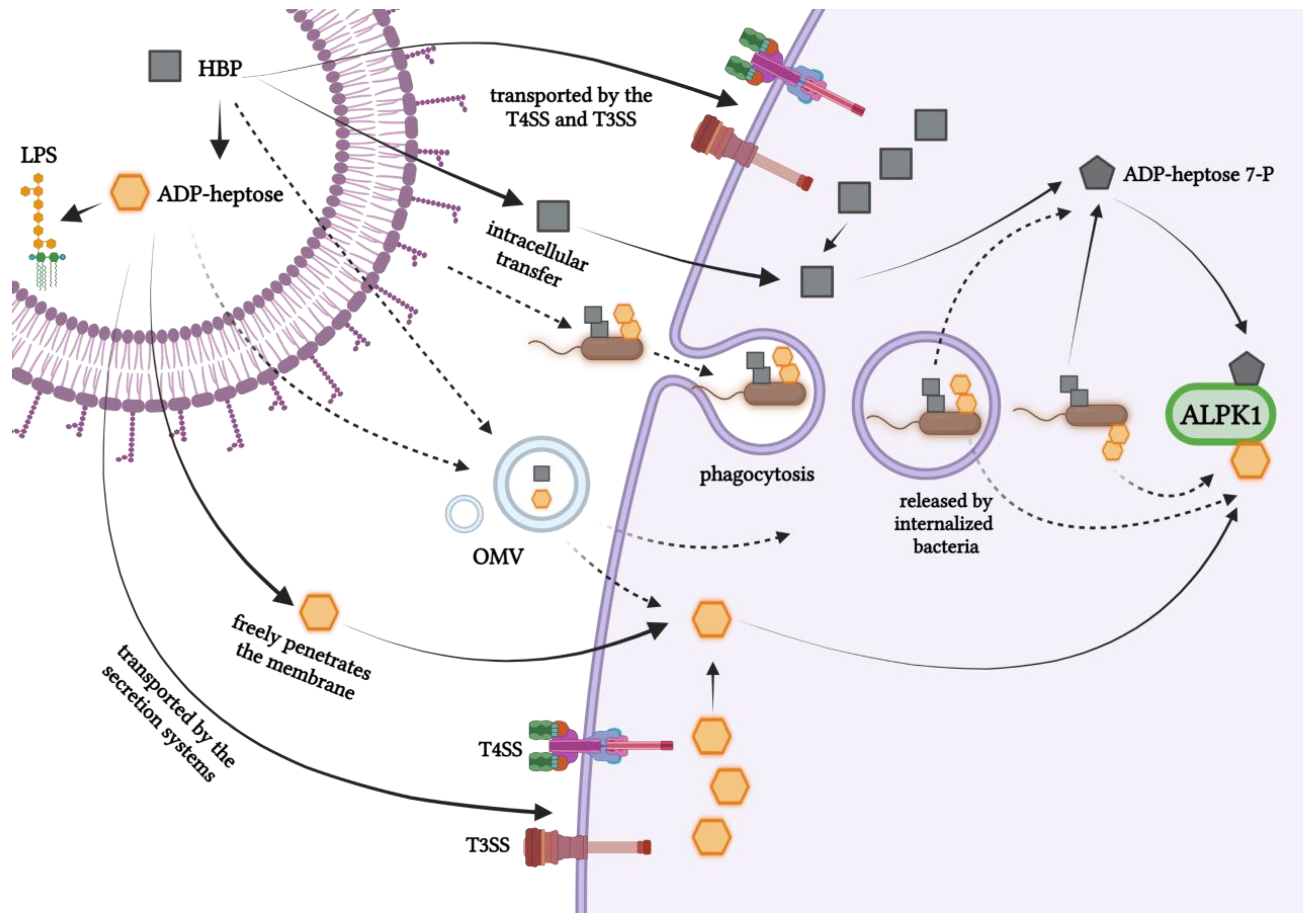
7. Intracellular Entry Routes of Bacterial Heptose
8. Puzzling Aspects of ADP-Heptose and ALPK1 in Health and Disease
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP-heptose | ADP-L-glycero-β-D-manno-heptose |
| HBP | D-glycero-β-D-manno-heptose 1,7-phosphate |
| HMP | D-glycero-β-D-manno-heptose 1-phosphate |
| Kdo | 3-Deoxy-D-manno-oct-2-ulosonic acid |
| LPS | Lipopolysaccharide |
| LOS | Lipooligosacharide |
| TLR4 | Toll-like receptor 4 |
| PAMP | Pathogen-associated molecular pattern |
| PRR | Pattern recognition receptor |
| TIFA | TRAF-interacting protein with a forkhead-associated domain |
| ALPK1 | Alpha kinase-1 |
| MyD88 | Myeloid differentiation primary response gene 88 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| IRF3 | Interferon regulatory factor 3 |
References
- Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 27 February 2017).
- Infectious Diseases Society of America. No ESKAPE! New Drugs against MRSA, Other Superbugs Still Lacking. ScienceDaily, 9 December 2008. [Google Scholar]
- Cui, J.; Duizer, C.; Bouwman, L.I.; van Rooijen, K.S.; Voogdt, C.G.P.; van Putten, J.P.M.; de Zoete, M.R. The ALPK1 pathway drives the inflammatory response to Campylobacter jejuni in human intestinal epithelial cells. PLoS Pathog. 2021, 17, e1009787. [Google Scholar] [CrossRef]
- Zhou, P.; She, Y.; Dong, N.; Li, P.; He, H.; Borio, A.; Wu, Q.; Lu, S.; Ding, X.; Cao, Y.; et al. Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature 2018, 561, 122–126. [Google Scholar] [CrossRef] [PubMed]
- García-Weber, D.; Dangeard, A.S.; Cornil, J.; Thai, L.; Rytter, H.; Zamyatina, A.; Mulard, L.A.; Arrieumerlou, C. ADP-heptose is a newly identified pathogen-associated molecular pattern of Shigella flexneri. EMBO Rep. 2018, 19, e46943. [Google Scholar] [CrossRef]
- Pfannkuch, L.; Hurwitz, R.; Trauisen, J.; Sigulla, J.; Poeschke, M.; Matzner, L.; Kosma, P.; Schmid, M.; Meyer, T.F. ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. FASEB J. 2019, 33, 9087–9099. [Google Scholar] [CrossRef]
- Holmes, C.L.; Smith, S.N.; Gurczynski, S.J.; Severin, G.B.; Unverdorben, L.V.; Vornhagen, J.; Mobley, H.L.T.; Bachman, M.A. The ADP-Heptose Biosynthesis Enzyme GmhB is a Conserved Gram-Negative Bacteremia Fitness Factor. Infect. Immun. 2022, 90, e0022422. [Google Scholar] [CrossRef]
- Milivojevic, M.; Dangeard, A.-S.; Kasper, C.A.; Tschon, T.; Emmenlauer, M.; Pique, C.; Schnupf, P.; Guignot, J.; Arrieumerlou, C. ALPK1 controls TIFA/TRAF6-dependent innate immunity against heptose-1,7-bisphosphate of gram-negative bacteria. PLoS Pathog. 2017, 13, e1006224. [Google Scholar] [CrossRef]
- Zimmermann, S.; Pfannkuch, L.; Al-Zeer, M.A.; Bartfeld, S.; Koch, M.; Liu, J.; Rechner, C.; Soerensen, M.; Sokolova, O.; Zamyatina, A.; et al. ALPK1- and TIFA-Dependent Innate Immune Response Triggered by the Helicobacter pylori Type IV Secretion System. Cell Rep. 2017, 20, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.K.; Leatham, M.P.; Conway, T.; Nuijten, P.J.M.; de Haan, L.A.M.; Krogfelt, K.A.; Cohen, P.S. An Escherichia coli MG1655 Lipopolysaccharide Deep-Rough Core Mutant Grows and Survives in Mouse Cecal Mucus but Fails to Colonize the Mouse Large Intestine. Infect. Immun. 2003, 71, 2142–2152. [Google Scholar] [CrossRef]
- De Leon, G.P.; Elowe, N.H.; Koteva, K.P.; Valvano, M.A.; Wright, G.D. An in vitro Screen of Bacterial Lipopolysaccharide Biosynthetic Enzymes Identifies an Inhibitor of ADP-Heptose Biosynthesis. Chem. Biol. 2006, 13, 437–441. [Google Scholar] [CrossRef]
- Matsuura, M. Structural Modifications of Bacterial Lipopolysaccharide That Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shao, F. Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr. Opin. Microbiol. 2016, 29, 37–42. [Google Scholar] [CrossRef]
- Raetz, C.R.H. Biochemistry of Endotoxins. Annu. Rev. Biochem. 1990, 59, 129–170. [Google Scholar] [CrossRef]
- Frirdich, E.; Whitfield, C. Lipopolysaccharide Inner Core Oligosaccharide Structure and Outer Membrane Stability in Human Pathogens Belonging to the Enterobacteriaceae. J. Endotoxin Res. 2005, 11, 133–144. [Google Scholar]
- Caroff, M.; Karibian, D.; Cavaillon, J.-M.; Haeffner-Cavaillon, N. Structural and functional analyses of bacterial lipopolysaccharides. Microbes Infect. 2002, 4, 915–926. [Google Scholar] [CrossRef]
- Süsskind, M.; Lindner, B.; Weimar, T.; Brade, H.; Holst, O. The structure of the lipopolysaccharide from Klebsiella oxytoca rough mutant R29 (O1-/K29-). Carbohydr. Res. 1998, 312, 91–95. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Kocharova, N.A.; Hynes, S.; Widmalm, G.; Andersen, L.P.; Jansson, P.-E.; Moran, A.P. Structural studies on lipopolysaccharides of serologically non-typable strains of Helicobacter pylori, AF1 and 007, expressing Lewis antigenic determinants. Eur. J. Biochem. 1999, 266, 123–131. [Google Scholar] [CrossRef]
- Rittig, M.G.; Kaufmann, A.; Robins, A.; Shaw, B.; Sprenger, H.; Gemsa, D.; Foulongne, V.; Rouot, B.; Dornand, J. Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intracellular trafficking and cytokine/chemokine release in human monocytes. J. Leukoc. Biol. 2003, 74, 1045–1055. [Google Scholar] [CrossRef]
- Moran, A.P.; Prendergast, M.M.; Appelmelk, B.J. Molecular mimicry of host structures by bacterial lipopolysaccharides and its contribution to disease. FEMS Immunol. Med. Microbiol. 1996, 16, 105–115. [Google Scholar] [CrossRef]
- Kilár, A.; Dörnyei, Á.; Kocsis, B. Structural characterization of bacterial lipopolysaccharides with mass spectrometry and on- and off-line separation techniques. Mass Spectrom. Rev. 2013, 32, 90–117. [Google Scholar] [CrossRef]
- Fregolino, E.; Fugazza, G.; Galano, E.; Gargiulo, V.; Landini, P.; Lanzetta, R.; Lindner, B.; Pagani, L.; Parrilli, M.; Holst, O.; et al. Complete Lipooligosaccharide Structure of the Clinical Isolate Acinetobacter baumannii, Strain SMAL. Eur. J. Org. Chem. 2010, 2010, 1345–1352. [Google Scholar] [CrossRef]
- Powers, M.J.; Trent, M.S. Expanding the paradigm for the outer membrane: Acinetobacter baumannii in the absence of endotoxin. Mol. Microbiol. 2017, 107, 47–56. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, W.; Zhang, B.; Liao, C.; Shao, Y. Characterization of a biofilm-forming Shigella flexneri phenotype due to deficiency in Hep biosynthesis. PeerJ 2016, 4, e2178. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a Molecule that Confers Lipopolysaccharide Responsiveness on Toll-like Receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Gioannini, T.; Teghanemt, A.; Zhang, D.; Levis, E.; Weiss, J. Monomeric endotoxin:protein complexes are essential for TLR4-dependent cell activation. J. Endotoxin Res. 2005, 11, 117–123. [Google Scholar] [CrossRef]
- Gioannini, T.L.; Teghanemt, A.; Zhang, D.; Coussens, N.P.; Dockstader, W.; Ramaswamy, S.; Weiss, J.P. Isolation of an endotoxin–MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef]
- Zamyatina, A.; Heine, H. Lipopolysaccharide Recognition in the Crossroads of TLR4 and Caspase-4/11 Mediated Inflammatory Pathways. Front. Immunol. 2020, 11, 585146. [Google Scholar] [CrossRef]
- Skirecki, T.; Cavaillon, J.-M. Inner sensors of endotoxin–implications for sepsis research and therapy. FEMS Microbiol. Rev. 2019, 43, 239–256. [Google Scholar] [CrossRef]
- Stein, S.C.; Faber, E.; Bats, S.H.; Murillo, T.; Speidel, Y.; Coombs, N.; Josenhans, C. Helicobacter pylori modulates host cell responses by CagT4SS-dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog. 2017, 13, e1006514. [Google Scholar] [CrossRef]
- García-Weber, D.; Arrieumerlou, C. ADP-heptose: A bacterial PAMP detected by the host sensor ALPK1. Cell. Mol. Life Sci. 2021, 78, 17–29. [Google Scholar] [CrossRef]
- Valvano, M.A.; Messner, P.; Kosma, P. Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology 2002, 148, 1979–1989. [Google Scholar] [CrossRef]
- Kneidinger, B.; Marolda, C.; Graninger, M.; Zamyatina, A.; McArthur, F.; Kosma, P.; Valvano, M.A.; Messner, P. Biosynthesis pathway of ADP-l-glycero-β-d-manno-heptose in Escherichia coli. J. Bacteriol. 2002, 184, 363–369. [Google Scholar] [CrossRef]
- Desroy, N.; Moreau, F.; Briet, S.; Le Fralliec, G.; Floquet, S.; Durant, L.; Vongsouthi, V.; Gerusz, V.; Denis, A.; Escaich, S. Towards Gram-negative antivirulence drugs: New inhibitors of HldE kinase. Bioorganic Med. Chem. 2009, 17, 1276–1289. [Google Scholar] [CrossRef]
- Gronow, S.; Brabetz, W.; Brade, H. Comparative functional characterization in vitro of heptosyltransferase I (WaaC) and II (WaaF) from Escherichia coli. Eur. J. Biochem. 2000, 267, 6602–6611. [Google Scholar] [CrossRef]
- Tang, W.; Guo, Z.; Cao, Z.; Wang, M.; Li, P.; Meng, X.; Zhao, X.; Xie, Z.; Wang, W.; Zhou, A.; et al. d-Sedoheptulose-7-phosphate is a common precursor for the heptoses of septacidin and hygromycin B. Proc. Natl. Acad. Sci. USA 2018, 115, 2818–2823. [Google Scholar] [CrossRef]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J.; Schindler, B.D.; Jacinto, P.; Kaatz, G.W.; Perrin, E.; Maggini, V.; et al. Pharmacology of polymyxins: New insights into an ‘old’ class of antibiotics. Futur. Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef]
- El-Sayed Ahmed, M.A.E.G.; Zhong, L.-L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.-B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef]
- Bergen, P.J.; Tsuji, B.T.; Bulitta, J.B.; Forrest, A.; Jacob, J.; Sidjabat, H.E.; Paterson, D.L.; Nation, R.L.; Li, J. Synergistic Killing of Multidrug-Resistant Pseudomonas aeruginosa at Multiple Inocula by Colistin Combined with Doripenem in an In Vitro Pharmacokinetic/Pharmacodynamic Model. Antimicrob. Agents Chemother. 2011, 55, 5685–5695. [Google Scholar] [CrossRef]
- Jernigan, M.G.; Press, E.G.; Nguyen, M.H.; Clancy, C.J.; Shields, R.K. The Combination of Doripenem and Colistin Is Bactericidal and Synergistic against Colistin-Resistant, Carbapenemase-Producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2012, 56, 3395–3398. [Google Scholar] [CrossRef]
- Oleksiuk, L.M.; Nguyen, M.H.; Press, E.G.; Updike, C.L.; O’Hara, J.A.; Doi, Y.; Clancy, C.J.; Shields, R.K. In Vitro Responses of Acinetobacter baumannii to Two- and Three-Drug Combinations following Exposure to Colistin and Doripenem. Antimicrob. Agents Chemother. 2014, 58, 1195–1199. [Google Scholar] [CrossRef]
- Ly, N.S.; Bulitta, J.B.; Rao, G.G.; Landersdorfer, C.B.; Holden, P.N.; Forrest, A.; Bergen, P.J.; Nation, R.L.; Li, J.; Tsuji, B.T. Colistin and doripenem combinations against Pseudomonas aeruginosa: Profiling the time course of synergistic killing and prevention of resistance. J. Antimicrob. Chemother. 2014, 70, 1434–1442. [Google Scholar] [CrossRef]
- Maifiah, M.H.M.; Creek, D.J.; Nation, R.L.; Forrest, A.; Tsuji, B.T.; Velkov, T.; Li, J. Untargeted metabolomics analysis reveals key pathways responsible for the synergistic killing of colistin and doripenem combination against Acinetobacter baumannii. Sci. Rep. 2017, 7, 45527. [Google Scholar] [CrossRef]
- Malott, R.J.; Keller, B.O.; Gaudet, R.G.; McCaw, S.E.; Lai, C.C.L.; Dobson-Belaire, W.N.; Hobbs, J.L.; Michael, F.S.; Cox, A.D.; Moraes, T.F.; et al. Neisseria gonorrhoeae- derived heptose elicits an innate immune response and drives HIV-1 expression. Proc. Natl. Acad. Sci. USA 2013, 110, 10234–10239. [Google Scholar] [CrossRef]
- Gaudet, R.G.; Sintsova, A.; Buckwalter, C.M.; Leung, N.; Cochrane, A.; Li, J.; Cox, A.D.; Moffat, J.; Gray-Owen, S.D. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 2015, 348, 1251–1255. [Google Scholar] [CrossRef]
- Gall, A.; Gaudet, R.G.; Gray-Owen, S.D.; Salama, N.R. TIFA Signaling in Gastric Epithelial Cells Initiates the cag Type 4 Secretion System-Dependent Innate Immune Response to Helicobacter pylori Infection. mBio 2017, 8, e01168-17. [Google Scholar] [CrossRef] [PubMed]
- Adekoya, I.A.; Guo, C.X.; Gray-Owen, S.D.; Cox, A.D.; Sauvageau, J. d-Glycero-β-d-Manno-Heptose 1-Phosphate and d-Glycero-β-d-Manno-Heptose 1,7-Biphosphate Are Both Innate Immune Agonists. J. Immunol. 2018, 201, 2385–2391. [Google Scholar] [CrossRef]
- Snelling, T.; Shpiro, N.; Gourlay, R.; Lamoliatte, F.; Cohen, P. Co-ordinated control of the ADP-heptose/ALPK1 signalling network by the E3 ligases TRAF6, TRAF2/c-IAP1 and LUBAC. Biochem. J. 2022, 479, 2195–2216. [Google Scholar] [CrossRef]
- Heine, M.; Cramm-Behrens, C.I.; Ansari, A.; Chu, H.-P.; Ryazanov, A.G.; Naim, H.Y.; Jacob, R. α-Kinase 1, a New Component in Apical ProteinTransport. J. Biol. Chem. 2005, 280, 25637–25643. [Google Scholar] [CrossRef]
- Lee, C.-P.; Chiang, S.-L.; Ko, A.M.-S.; Liu, Y.-F.; Ma, C.; Lu, C.-Y.; Huang, C.-M.; Chang, J.-G.; Kuo, T.-M.; Chen, C.-L.; et al. ALPK1 phosphorylates myosin IIA modulating TNF-α trafficking in gout flares. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Rashid, M.; van der Horst, M.; Mentzel, T.; Butera, F.; Ferreira, I.; Pance, A.; Rütten, A.; Luzar, B.; Marusic, Z.; Aubain, N.D.S.; et al. ALPK1 hotspot mutation as a driver of human spiradenoma and spiradenocarcinoma. Nat. Commun. 2019, 10, 2213. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Azzarà, A.; Molinario, C.; Pietrobono, R.; Rigante, D.; Verrecchia, E.; Sicignano, L.L.; Genuardi, M.; Gurrieri, F.; Manna, R. Rare missense variants in the ALPK1 gene may predispose to periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) syndrome. Eur. J. Hum. Genet. 2019, 27, 1361–1368. [Google Scholar] [CrossRef]
- Williams, L.B.; Javed, A.; Sabri, A.; Morgan, D.J.; Huff, C.D.; Grigg, J.R.; Heng, X.T.; Khng, A.J.; Hollink, I.H.I.M.; Morrison, M.A.; et al. ALPK1 missense pathogenic variant in five families leads to ROSAH syndrome, an ocular multisystem autosomal dominant disorder. Genet. Med. 2019, 21, 2103–2115. [Google Scholar] [CrossRef]
- Yamada, Y.; Matsui, K.; Takeuchi, I.; Oguri, M.; Fujimaki, T. Association of genetic variants of the α-kinase 1 gene with type 2 diabetes mellitus in a longitudinal population-based genetic epidemiological study. Biomed. Rep. 2015, 3, 347–354. [Google Scholar] [CrossRef]
- Hu, X.; Yang, C.; Wang, P.G.; Zhang, G.-L. ADP-heptose: A new innate immune modulator. Carbohydr. Res. 2019, 473, 123–128. [Google Scholar] [CrossRef]
- Kanamori, M.; Suzuki, H.; Saito, R.; Muramatsu, M.; Hayashizaki, Y. T2BP, a Novel TRAF2 Binding Protein, Can Activate NF-κB and AP-1 without TNF Stimulation. Biochem. Biophys. Res. Commun. 2002, 290, 1108–1113. [Google Scholar] [CrossRef]
- Takatsuna, H.; Kato, H.; Gohda, J.; Akiyama, T.; Moriya, A.; Okamoto, Y.; Yamagata, Y.; Otsuka, M.; Umezawa, K.; Semba, K.; et al. Identification of TIFA as an Adapter Protein That Links Tumor Necrosis Factor Receptor-associated Factor 6 (TRAF6) to Interleukin-1 (IL-1) Receptor-associated Kinase-1 (IRAK-1) in IL-1 Receptor Signaling. J. Biol. Chem. 2003, 278, 12144–12150. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.-K.; Sun, L.; Inoue, J.-I.; Chen, Z.J. TIFA activates IκB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6. Proc. Natl. Acad. Sci. USA 2004, 101, 15318–15323. [Google Scholar] [CrossRef] [PubMed]
- García-Weber, D.; Dangeard, A.-S.; Teixeira, V.; Hauke, M.; Carreaux, A.; Josenhans, C.; Arrieumerlou, C. In vitro ALPK1 Kinase Assay Reveals New Insights into ADP-Heptose Sensing Pathway and Kinase Activity of Disease-Associated ALPK1 Mutants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gaudet, R.G.; Guo, C.X.; Molinaro, R.; Kottwitz, H.; Rohde, J.R.; Dangeard, A.-S.; Arrieumerlou, C.; Girardin, S.E.; Gray-Owen, S.D. Innate Recognition of Intracellular Bacterial Growth Is Driven by the TIFA-Dependent Cytosolic Surveillance Pathway. Cell Rep. 2017, 19, 1418–1430. [Google Scholar] [CrossRef]
- Carson, D.; Barry, R.; Hopkins, E.G.; Roumeliotis, T.I.; García-Weber, D.; Mullineaux-Sanders, C.; Elinav, E.; Arrieumerlou, C.; Choudhary, J.S.; Frankel, G. Citrobacter rodentium induces rapid and unique metabolic and inflammatory responses in mice suffering from severe disease. Cell. Microbiol. 2020, 22, e13126. [Google Scholar] [CrossRef] [PubMed]
- Faass, L.; Stein, S.C.; Hauke, M.; Gapp, M.; Albanese, M.; Josenhans, C. Contribution of Heptose Metabolites and the cag Pathogenicity Island to the Activation of Monocytes/Macrophages by Helicobacter pylori. Front. Immunol. 2021, 12, 632154. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, J.; Franchi, L.; Backert, S.; Núñez, G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1β production in Helicobacter pylori infected dendritic cells. Eur. J. Immunol. 2013, 43, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Ballhorn, W.; Voland, P.; Eisenächer, K.; Mages, J.; Rad, L.; Ferstl, R.; Lang, R.; Wagner, H.; Schmid, R.M.; et al. Extracellular and Intracellular Pattern Recognition Receptors Cooperate in the Recognition of Helicobacter pylori. Gastroenterology 2009, 136, 2247–2257. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Wei, T.-Y.W.; Li, S.; Wang, S.-C.; He, M.; Martin, M.; Zhang, J.; Shentu, T.-P.; Xiao, H.; Kang, J.; et al. TIFA as a crucial mediator for NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2016, 113, 15078–15083. [Google Scholar] [CrossRef]
- Chiu, S.-F.; Teng, K.-W.; Wang, P.-C.; Chung, H.-Y.; Wang, C.-J.; Cheng, H.-C.; Kao, M.-C. Helicobacter pylori GmhB enzyme involved in ADP-heptose biosynthesis pathway is essential for lipopolysaccharide biosynthesis and bacterial virulence. Virulence 2021, 12, 1610–1628. [Google Scholar] [CrossRef]
- Beveridge, T.J. Structures of Gram-Negative Cell Walls and Their Derived Membrane Vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef]
- Yu, C.-K.; Wang, C.-J.; Chew, Y.; Wang, P.-C.; Yin, H.-S.; Kao, M.-C. Functional characterization of Helicobacter pylori 26695 sedoheptulose 7-phosphate isomerase encoded by hp0857 and its association with lipopolysaccharide biosynthesis and adhesion. Biochem. Biophys. Res. Commun. 2016, 477, 794–800. [Google Scholar] [CrossRef]
- Chang, P.-C.; Wang, C.-J.; You, C.-K.; Kao, M.-C. Effects of a HP0859 (rfaD) knockout mutation on lipopolysaccharide structure of Helicobacter pylori 26695 and the bacterial adhesion on AGS cells. Biochem. Biophys. Res. Commun. 2011, 405, 497–502. [Google Scholar] [CrossRef]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Mentucci, F.; Voli, A.; Petrella, A.; Porta, A.; Tosco, A. Helicobacter pylori Pathogen-Associated Molecular Patterns: Friends or Foes? Int. J. Mol. Sci. 2022, 23, 3531. [Google Scholar] [CrossRef]
- Auerbuch, V.; Golenbock, D.T.; Isberg, R.R. Innate Immune Recognition of Yersinia pseudotuberculosis Type III Secretion. PLoS Pathog. 2009, 5, e1000686. [Google Scholar] [CrossRef]
- Bruno, V.M.; Hannemann, S.; Lara-Tejero, M.; Flavell, R.A.; Kleinstein, S.H.; Galán, J.E. Salmonella Typhimurium Type III Secretion Effectors Stimulate Innate Immune Responses in Cultured Epithelial Cells. PLoS Pathog. 2009, 5, e1000538. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Sharon, S.; Hyams, M.; Zhang, L.; Kobi, S.; Katsowich, N.; Dishon, S.; Nussbaum, G.; Dong, N.; Shao, F.; et al. Epithelial cells detect functional type III secretion system of enteropathogenic Escherichia coli through a novel NF-κB signaling pathway. PLoS Pathog. 2017, 13, e1006472. [Google Scholar] [CrossRef] [PubMed]
- Hii, C.; Sun, G.W.; Goh, J.W.K.; Lu, J.; Stevens, M.P.; Gan, Y. Interleukin-8 Induction by Burkholderia pseudomallei Can Occur without Toll-Like Receptor Signaling but Requires a Functional Type III Secretion System. J. Infect. Dis. 2008, 197, 1537–1547. [Google Scholar] [CrossRef]
- Jan, A.T. Outer Membrane Vesicles (OMVs) of Gram-negative Bacteria: A Perspective Update. Front. Microbiol. 2017, 8, 1053. [Google Scholar] [CrossRef]
- Ryzhakov, G.; West, N.R.; Franchini, F.; Clare, S.; Ilott, N.E.; Sansom, S.N.; Bullers, S.J.; Pearson, C.; Costain, A.; Vaughan-Jackson, A.; et al. Alpha kinase 1 controls intestinal inflammation by suppressing the IL-12/Th1 axis. Nat. Commun. 2018, 9, 3797. [Google Scholar] [CrossRef]
- Bauer, M.; Nascakova, Z.; Mihai, A.-I.; Cheng, P.F.; Levesque, M.P.; Lampart, S.; Hurwitz, R.; Pfannkuch, L.; Dobrovolna, J.; Jacobs, M.; et al. The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat. Commun. 2020, 11, 5117. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Zheng, S.; Li, M.; Xu, C.; Jia, D.; Qi, Y.; Hou, T.; Wang, L.; Wang, B.; et al. Fusobacterium nucleatum promotes colorectal cancer cells adhesion to endothelial cells and facilitates extravasation and metastasis by inducing ALPK1/NF-κB/ICAM1 axis. Gut Microbes 2022, 14, 2038852. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-K.; Hua, C.-H.; Hsu, H.-T.; Kuo, T.-M.; Chung, C.-M.; Lee, C.-P.; Tsai, M.-H.; Yeh, K.-T.; Ko, Y.-C. ALPK1 Expression Is Associated with Lymph Node Metastasis and Tumor Growth in Oral Squamous Cell Carcinoma Patients. Am. J. Pathol. 2019, 189, 190–199. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Garcia-Weber, D.; Lashermes, A.; Larraufie, P.; Marinelli, L.; Teixeira, V.; Rolland, A.; Béguet-Crespel, F.; Brochard, V.; Quatremare, T.; et al. Akkermansia muciniphila upregulates genes involved in maintaining the intestinal barrier function via ADP-heptose-dependent activation of the ALPK1/TIFA pathway. Gut Microbes 2022, 14, 2110639. [Google Scholar] [CrossRef] [PubMed]
- Kozycki, C.T.; Kodati, S.; Huryn, L.; Wang, H.; Warner, B.M.; Jani, P.; Hammoud, D.; Abu-Asab, M.S.; Jittayasothorn, Y.; Mattapallil, M.J.; et al. Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: Functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome. Ann. Rheum. Dis. 2022, 81, 1453–1464. [Google Scholar] [CrossRef]
- Yamada, Y.; Matsui, K.; Takeuchi, I.; Fujimaki, T. Association of genetic variants with dyslipidemia and chronic kidney disease in a longitudinal population-based genetic epidemiological study. Int. J. Mol. Med. 2015, 35, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.M.-S.; Tu, H.-P.; Liu, T.-T.; Chang, J.-G.; Yuo, C.-Y.; Chiang, S.-L.; Chang, S.-J.; Liu, Y.-F.; Ko, A.M.-J.; Lee, C.-H.; et al. ALPK1 genetic regulation and risk in relation to gout. Int. J. Epidemiol. 2013, 42, 466. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.-F.; Lee, H.-H.; Chang, Y.-S.; Lin, C.-L.; Liu, T.-Y.; Chen, Y.-C.; Yen, J.-C.; Lee, Y.-T.; Lin, C.-Y.; Wu, S.-H.; et al. Down-regulated and Commonly mutated ALPK1 in Lung and Colorectal Cancers. Sci. Rep. 2016, 6, 27350. [Google Scholar] [CrossRef]
- Ko, A.M.-S.; Tu, H.-P.; Ko, Y.-C. Systematic Review of the Role of Alpha-Protein Kinase 1 in Cancer and Cancer-Related Inflammatory Diseases. Cancers 2022, 14, 4390. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Lim, M.C.C.; Sokolova, O.; Backert, S.; Meyer, T.F.; Naumann, M. TIFA has dual functions in Helicobacter pylori-induced classical and alternative NF-κB pathways. EMBO Rep. 2021, 22, e52878. [Google Scholar] [CrossRef]
- Kamoshida, G.; Akaji, T.; Takemoto, N.; Suzuki, Y.; Sato, Y.; Kai, D.; Hibino, T.; Yamaguchi, D.; Kikuchi-Ueda, T.; Nishida, S.; et al. Lipopolysaccharide-Deficient Acinetobacter baumannii Due to Colistin Resistance Is Killed by Neutrophil-Produced Lysozyme. Front. Microbiol. 2020, 11, 573. [Google Scholar] [CrossRef]
- Marszalik, A.; Sidor, K.; Kraśnicka, A.; Wróblewska, M.; Skirecki, T.; Jagielski, T.; Stachowiak, R. Acinetobacter baumannii–Virulence Factors and Epidemiology of Infections. Postępy Mikrobiol.-Adv. Microbiol. 2021, 60, 267–279. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidor, K.; Skirecki, T. A Bittersweet Kiss of Gram-Negative Bacteria: The Role of ADP-Heptose in the Pathogenesis of Infection. Microorganisms 2023, 11, 1316. https://doi.org/10.3390/microorganisms11051316
Sidor K, Skirecki T. A Bittersweet Kiss of Gram-Negative Bacteria: The Role of ADP-Heptose in the Pathogenesis of Infection. Microorganisms. 2023; 11(5):1316. https://doi.org/10.3390/microorganisms11051316
Chicago/Turabian StyleSidor, Karolina, and Tomasz Skirecki. 2023. "A Bittersweet Kiss of Gram-Negative Bacteria: The Role of ADP-Heptose in the Pathogenesis of Infection" Microorganisms 11, no. 5: 1316. https://doi.org/10.3390/microorganisms11051316