
Identification of Two Clusters in Renal Pelvis Urobiome of Unilateral Stone Formers Using 2bRAD-M


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Sample Collection and Processing
2.3. DNA Extraction, Library Preparation and Sequencing
2.4. Sequencing Processing and Quantitative Analysis
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
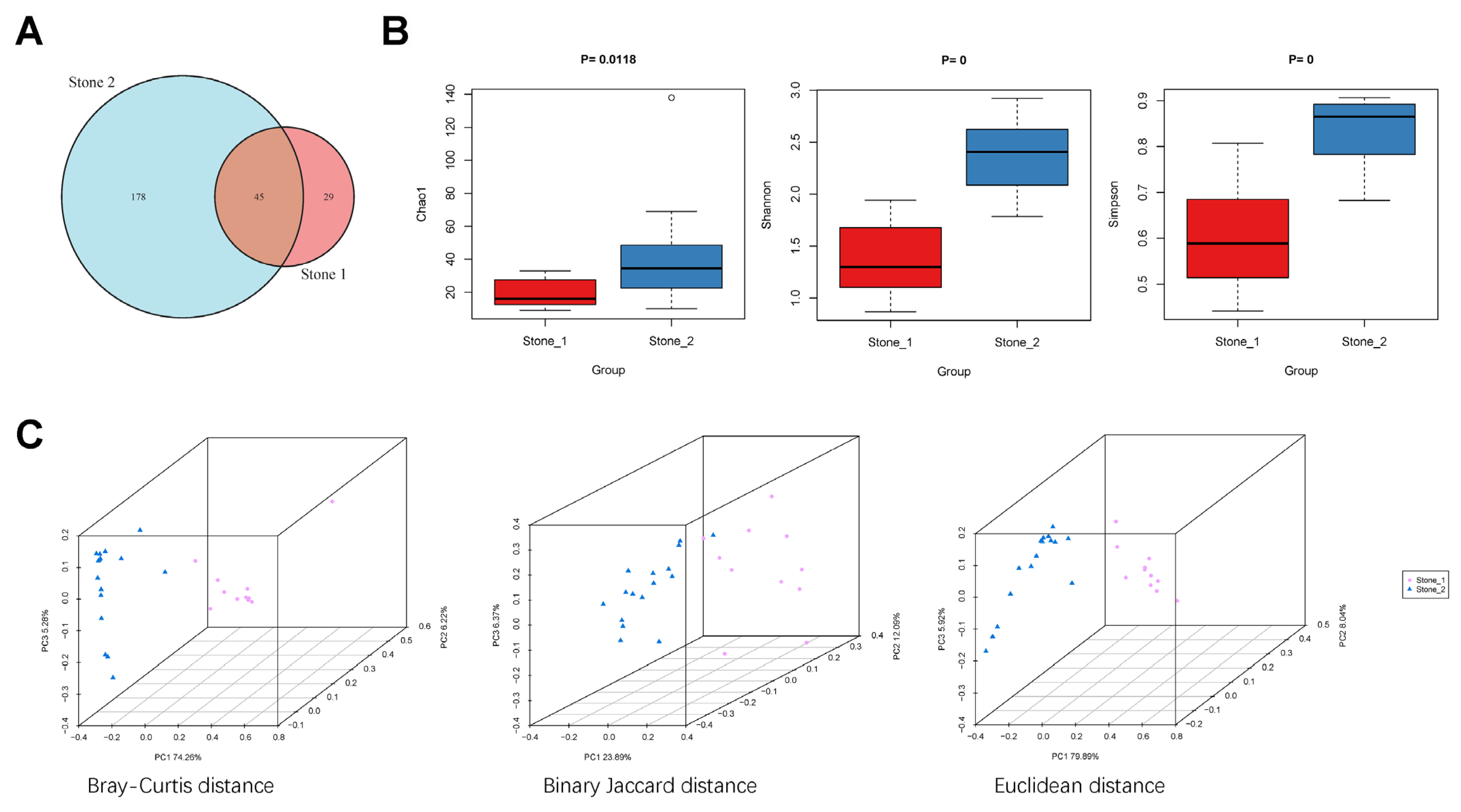
3.1. Clustering of the Renal Pelvis Urine Samples
3.2. Biodiversity of the Renal Pelvis Urobiome between Stone1 and Stone2
3.3. Bacterial Community Composition between Stone1 and Stone2
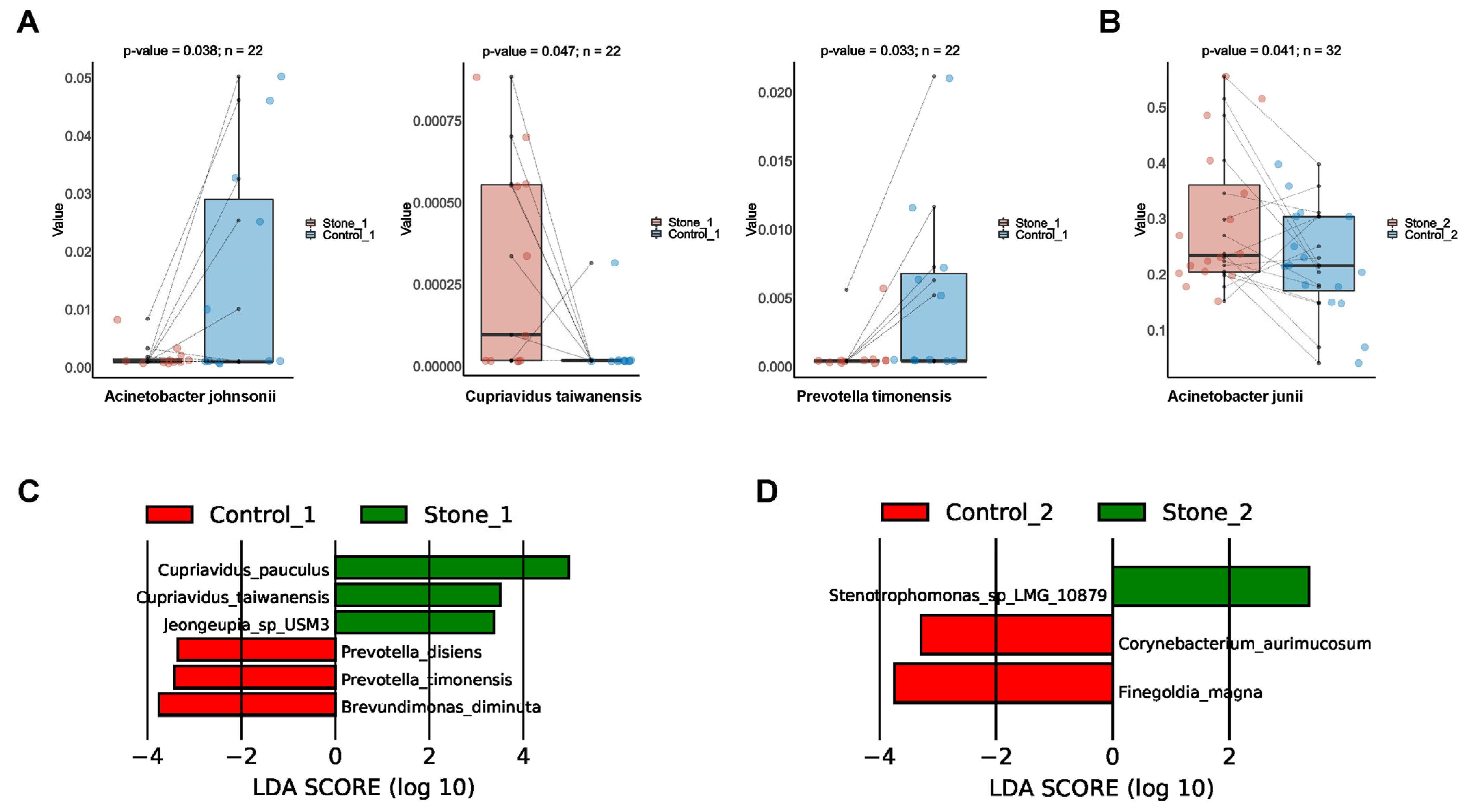
3.4. Differential Bacterial Taxa between Stone1 and Stone2
3.5. Classification of Two Clusters Using Random Forest Model
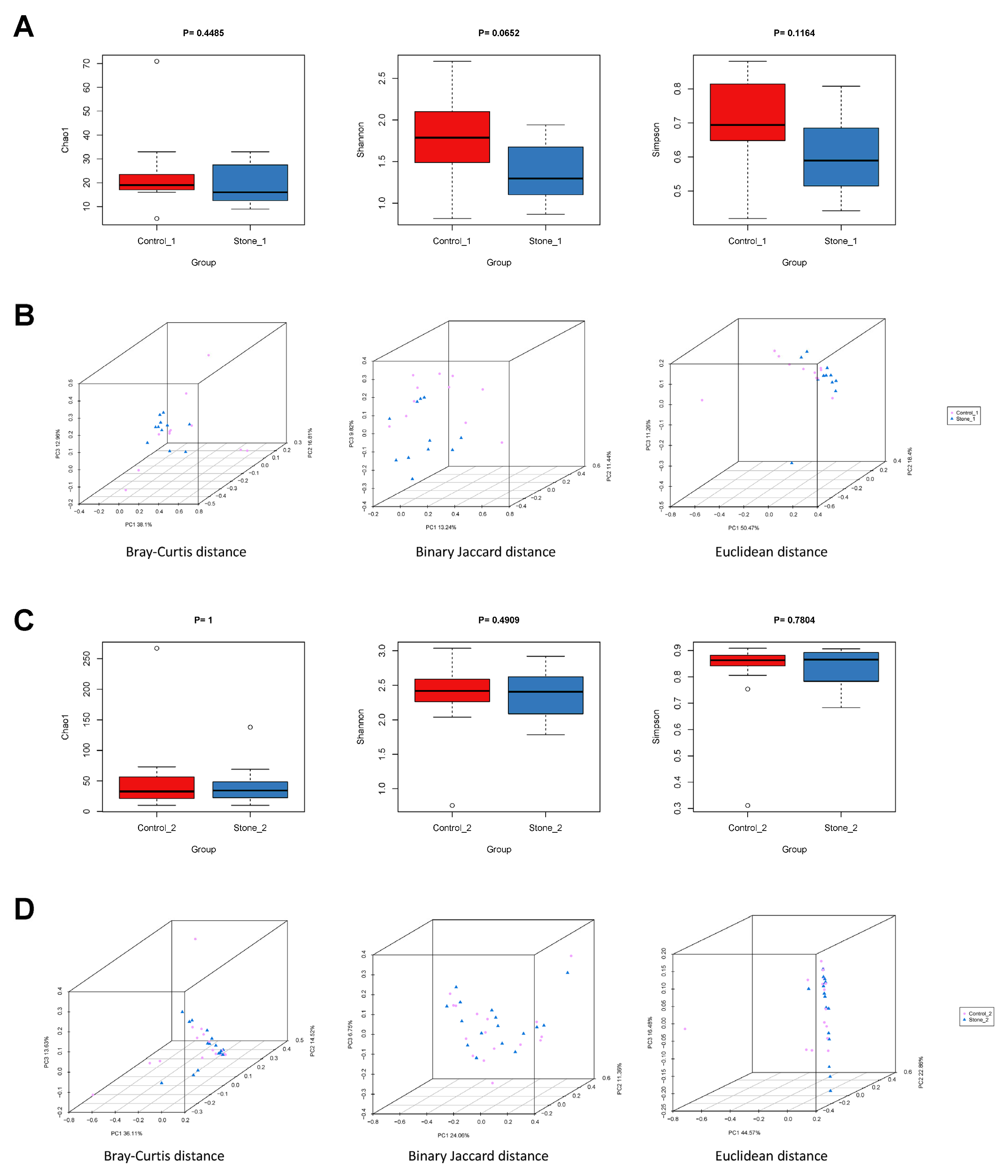
3.6. Biodiversity of the Renal Pelvis Urobiome between Stone1 and Control1/Stone2 and Control2
3.7. Differential Bacterial Taxa between Stone1 and Control1/Stone2 and Control2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeng, G.; Mai, Z.; Xia, S.; Wang, Z.; Zhang, K.; Wang, L.; Long, Y.; Ma, J.; Li, Y.; Wan, S.P.; et al. Prevalence of kidney stones in China: An ultrasonography based cross-sectional study. BJU Int. 2017, 120, 109–116. [Google Scholar] [CrossRef]
- D’Costa, M.R.; Haley, W.E.; Mara, K.C.; Enders, F.T.; Vrtiska, T.J.; Pais, V.M.; Jacobsen, S.J.; McCollough, C.H.; Lieske, J.C.; Rule, A.D. Symptomatic and Radiographic Manifestations of Kidney Stone Recurrence and Their Prediction by Risk Factors: A Prospective Cohort Study. J. Am. Soc. Nephrol. 2019, 30, 1251–1260. [Google Scholar] [CrossRef]
- Thongprayoon, C.; Krambeck, A.E.; Rule, A.D. Determining the true burden of kidney stone disease. Nat. Rev. Nephrol. 2020, 16, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Baumann, J.M.; Affolter, B. From crystalluria to kidney stones, some physicochemical aspects of calcium nephrolithiasis. World J. Nephrol. 2014, 3, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Xia, Q.D.; Yang, Y.Y.; Li, C.; Zhang, J.Q.; Xu, J.Z.; Qin, B.L.; Xun, Y.; Wang, S.G. The role of microbiome: A novel insight into urolithiasis. Crit. Rev. Microbiol. 2022, 49, 177–196. [Google Scholar] [CrossRef]
- Whiteside, S.A.; Razvi, H.; Dave, S.; Reid, G.; Burton, J.P. The microbiome of the urinary tract--a role beyond infection. Nat. Rev. Urol. 2015, 12, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Karam, A.; Mjaess, G.; Albisinni, S.; El Daccache, Y.; Farah, M.; Daou, S.; Kazzi, H.; Hassoun, R.; Bou Kheir, G.; Aoun, F.; et al. Uncovering the role of urinary microbiota in urological tumors: A systematic review of literature. World J. Urol. 2022, 40, 951–964. [Google Scholar] [CrossRef]
- Xie, J.; Huang, J.S.; Huang, X.J.; Peng, J.M.; Yu, Z.; Yuan, Y.Q.; Xiao, K.F.; Guo, J.N. Profiling the urinary microbiome in men with calcium-based kidney stones. BMC Microbiol. 2020, 20, 41. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, N.; Jiang, P.; Zhai, Q.; Li, C.; Yu, D.; Wu, Y.; Zhang, Y.; Lv, L.; Xu, X.; et al. Characteristics of the urinary microbiome in kidney stone patients with hypertension. J. Transl. Med. 2020, 18, 130. [Google Scholar] [CrossRef]
- Dornbier, R.A.; Bajic, P.; Van Kuiken, M.; Jardaneh, A.; Lin, H.; Gao, X.; Knudsen, B.; Dong, Q.; Wolfe, A.J.; Schwaderer, A.L. The microbiome of calcium-based urinary stones. Urolithiasis 2020, 48, 191–199. [Google Scholar] [CrossRef]
- Shen, C.; Zhu, Q.; Dong, F.; Wang, W.; Fan, B.; Li, K.; Chen, J.; Hu, S.; He, Z.; Li, X. Identifying Two Novel Clusters in Calcium Oxalate Stones With Urinary Tract Infection Using 16S rDNA Sequencing. Front. Cell. Infect. Microbiol. 2021, 11, 723781. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, S.; Zhu, P.; Tzehau, L.; Zhao, H.; Lv, J.; Zhang, R.; Zhou, L.; Niu, Q.; Wang, X.; et al. Species-resolved sequencing of low-biomass or degraded microbiomes using 2bRAD-M. Genome Biol. 2022, 23, 36. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, B.; Simpson, G.; Solymos, P.; Stevens, H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.2-1. Agric. Sci. 2015, 2, 1–2. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by RandomForest. Forest 2001, 2, 23. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Daskalakis, M.I.; Magoulas, A.; Kotoulas, G.; Catsikis, I.; Bakolas, A.; Karageorgis, A.P.; Mavridou, A.; Doulia, D.; Rigas, F. Pseudomonas, Pantoea and Cupriavidus isolates induce calcium carbonate precipitation for biorestoration of ornamental stone. J. Appl. Microbiol. 2013, 115, 409–423. [Google Scholar] [CrossRef]
- Daskalakis, M.I.; Magoulas, A.; Kotoulas, G.; Katsikis, I.; Bakolas, A.; Karageorgis, A.P.; Mavridou, A.; Doulia, D.; Rigas, F. Cupriavidus metallidurans biomineralization ability and its application as a bioconsolidation enhancer for ornamental marble stone. Appl. Microbiol. Biotechnol. 2014, 98, 6871–6883. [Google Scholar] [CrossRef]
- Liu, J.; Su, J.; Ali, A.; Wang, Z.; Zhang, R. Potential of a novel facultative anaerobic denitrifying Cupriavidus sp. W12 to remove fluoride and calcium through calcium bioprecipitation. J. Hazard. Mater. 2022, 423, 126976. [Google Scholar] [CrossRef]
- Randall, A. THE ORIGIN AND GROWTH OF RENAL CALCULI. Ann. Surg. 1937, 105, 1009–1027. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.L.; Gillen, D.L.; Williams, J.C., Jr.; Evan, A.P.; Bledsoe, S.B.; Coe, F.L.; Worcester, E.M.; Matlaga, B.R.; Munch, L.C.; Lingeman, J.E. A formal test of the hypothesis that idiopathic calcium oxalate stones grow on Randall’s plaque. BJU Int. 2009, 103, 966–971. [Google Scholar] [CrossRef]
- Gay, C.; Letavernier, E.; Verpont, M.C.; Walls, M.; Bazin, D.; Daudon, M.; Nassif, N.; Stéphan, O.; de Frutos, M. Nanoscale Analysis of Randall’s Plaques by Electron Energy Loss Spectromicroscopy: Insight in Early Biomineral Formation in Human Kidney. ACS Nano 2020, 14, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, N.; Wu, Y.; Jiang, P.; Jiang, T.; Wang, Y.; Zhang, Y.; Zhai, Q.; Zou, Y.; Feng, N. The pelvis urinary microbiome in patients with kidney stones and clinical associations. BMC Microbiol. 2020, 20, 336. [Google Scholar] [CrossRef]
- Tang, R.; Jiang, Y.; Tan, A.; Ye, J.; Xian, X.; Xie, Y.; Wang, Q.; Yao, Z.; Mo, Z. 16S rRNA gene sequencing reveals altered composition of gut microbiota in individuals with kidney stones. Urolithiasis 2018, 46, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Barr-Beare, E.; Saxena, V.; Hilt, E.E.; Thomas-White, K.; Schober, M.; Li, B.; Becknell, B.; Hains, D.S.; Wolfe, A.J.; Schwaderer, A.L. The Interaction between Enterobacteriaceae and Calcium Oxalate Deposits. PLoS ONE 2015, 10, e0139575. [Google Scholar] [CrossRef] [PubMed]
- Fishman, M.R.; Giglio, K.; Fay, D.; Filiatrault, M.J. Physiological and genetic characterization of calcium phosphate precipitation by Pseudomonas species. Sci. Rep. 2018, 8, 10156. [Google Scholar] [CrossRef]
- Safari, A.; Habimana, O.; Allen, A.; Casey, E. The significance of calcium ions on Pseudomonas fluorescens biofilms—A structural and mechanical study. Biofouling 2014, 30, 859–869. [Google Scholar] [CrossRef]
- Chutipongtanate, S.; Sutthimethakorn, S.; Chiangjong, W.; Thongboonkerd, V. Bacteria can promote calcium oxalate crystal growth and aggregation. J. Biol. Inorg. Chem. 2013, 18, 299–308. [Google Scholar] [CrossRef]
- Kanlaya, R.; Naruepantawart, O.; Thongboonkerd, V. Flagellum Is Responsible for Promoting Effects of Viable Escherichia coli on Calcium Oxalate Crystallization, Crystal Growth, and Crystal Aggregation. Front. Microbiol. 2019, 10, 2507. [Google Scholar] [CrossRef]
- Djojodimedjo, T.; Soebadi, D.M.; Soetjipto. Escherichia coli infection induces mucosal damage and expression of proteins promoting urinary stone formation. Urolithiasis 2013, 41, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Domrongkitchaiporn, S.; Stitchantrakul, W.; Kochakarn, W. Causes of hypocitraturia in recurrent calcium stone formers: Focusing on urinary potassium excretion. Am. J. Kidney Dis. 2006, 48, 546–554. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Stone1 (n = 11) | Stone2 (n = 16) | p-Value |
|---|---|---|---|
| Age | 51.09 ± 18.26 | 48.13 ± 14.55 | 0.643 |
| Gender | 0.692 | ||
| Male | 8 (72.7%) | 10 (62.5%) | |
| Female | 3 (27.3%) | 6 (37.5%) | |
| Body mass index (kg/m2) | 24.34 ± 3.53 | 23.78 ± 1.79 | 0.575 |
| Stone side | 0.097 | ||
| Left | 5 (45.5%) | 13 (81.3%) | |
| Right | 6 (54.5%) | 3 (18.8%) | |
| Stone count | 1.000 | ||
| Single | 7 (63.6%) | 9 (56.3%) | |
| Multiple | 4 (36.4%) | 7 (43.8%) | |
| Stone composition | 0.496 | ||
| CaOx | 3 (27.3%) | 4 (25%) | |
| CaOx + CaP | 1 (9.1%) | 5 (31.3%) | |
| Uric acid | 2 (18.2%) | 1 (6.3%) | |
| Not available | 5 (45.5%) | 6 (37.5%) | |
| Comorbidities | 0.242 | ||
| Hypertension | 1 (9.1%) | 5 (31.3%) | |
| Diabetes | 0 (0%) | 1 (6.3%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-Y.; Miao, L.-T.; Zhang, J.-Q.; Wang, S.-G. Identification of Two Clusters in Renal Pelvis Urobiome of Unilateral Stone Formers Using 2bRAD-M. Microorganisms 2023, 11, 2276. https://doi.org/10.3390/microorganisms11092276
Hong S-Y, Miao L-T, Zhang J-Q, Wang S-G. Identification of Two Clusters in Renal Pelvis Urobiome of Unilateral Stone Formers Using 2bRAD-M. Microorganisms. 2023; 11(9):2276. https://doi.org/10.3390/microorganisms11092276
Chicago/Turabian StyleHong, Sen-Yuan, Lin-Tao Miao, Jia-Qiao Zhang, and Shao-Gang Wang. 2023. "Identification of Two Clusters in Renal Pelvis Urobiome of Unilateral Stone Formers Using 2bRAD-M" Microorganisms 11, no. 9: 2276. https://doi.org/10.3390/microorganisms11092276
APA StyleHong, S.-Y., Miao, L.-T., Zhang, J.-Q., & Wang, S.-G. (2023). Identification of Two Clusters in Renal Pelvis Urobiome of Unilateral Stone Formers Using 2bRAD-M. Microorganisms, 11(9), 2276. https://doi.org/10.3390/microorganisms11092276