
Metagenomic of Liver Tissue Identified at Least Two Genera of Totivirus-like Viruses in Molossus molossus Bats

 , , , , ,
, , , , ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Processing of Samples
2.3. Nucleic Acid Extraction (DNA/RNA)
2.4. Preparation of Libraries for the Illumina Platform
2.5. Reads Trimming and Contig Classification
2.6. Genome Annotation
2.7. Genetic Distances
2.8. Phylogenetic Analysis
3. Results
3.1. Contigs Quality and BlastX Similarities
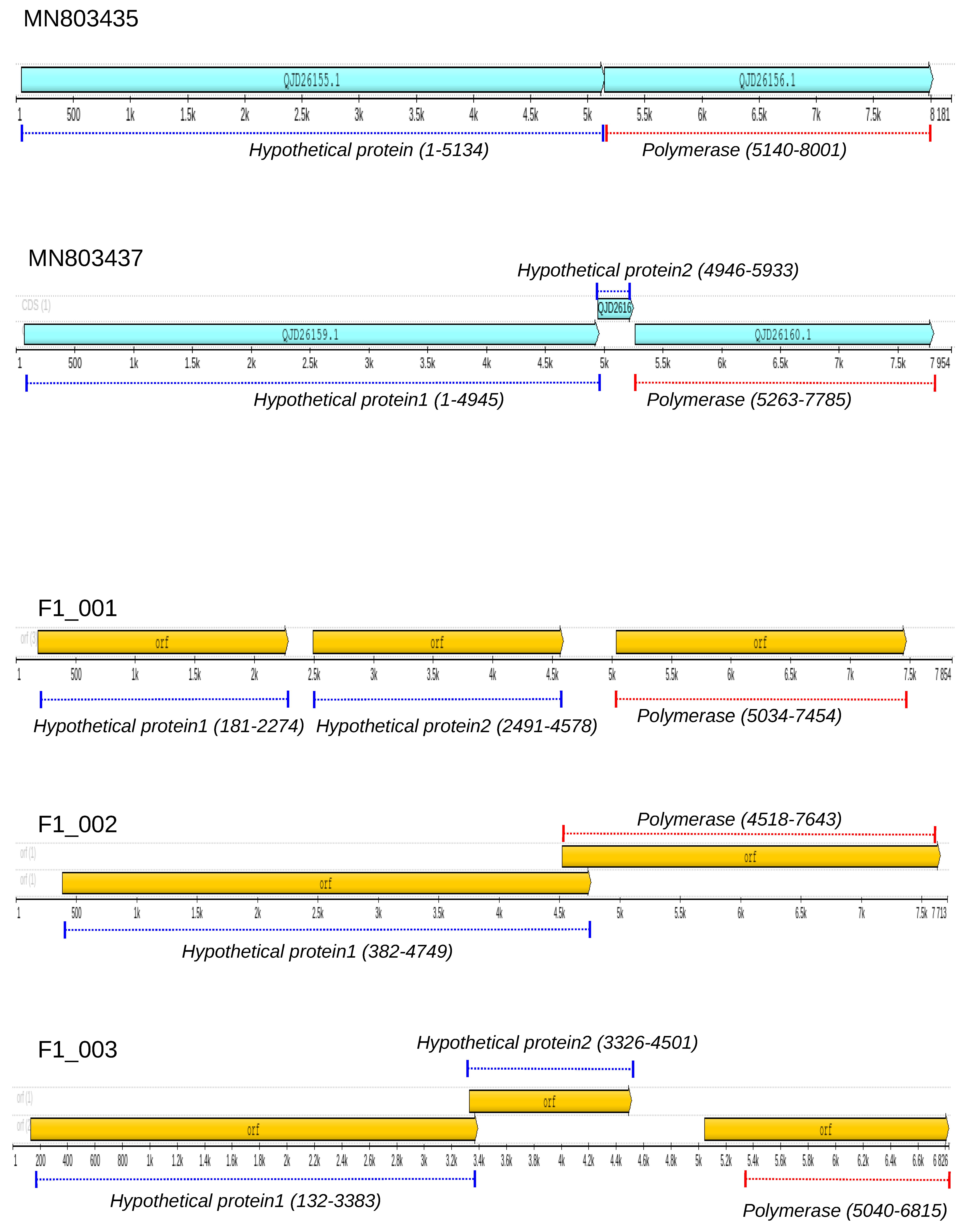
3.2. Genome Annotation of Totiviruses Identified in Bats
3.3. RNA-Dependent RNA-Polymerase of Totiviruses
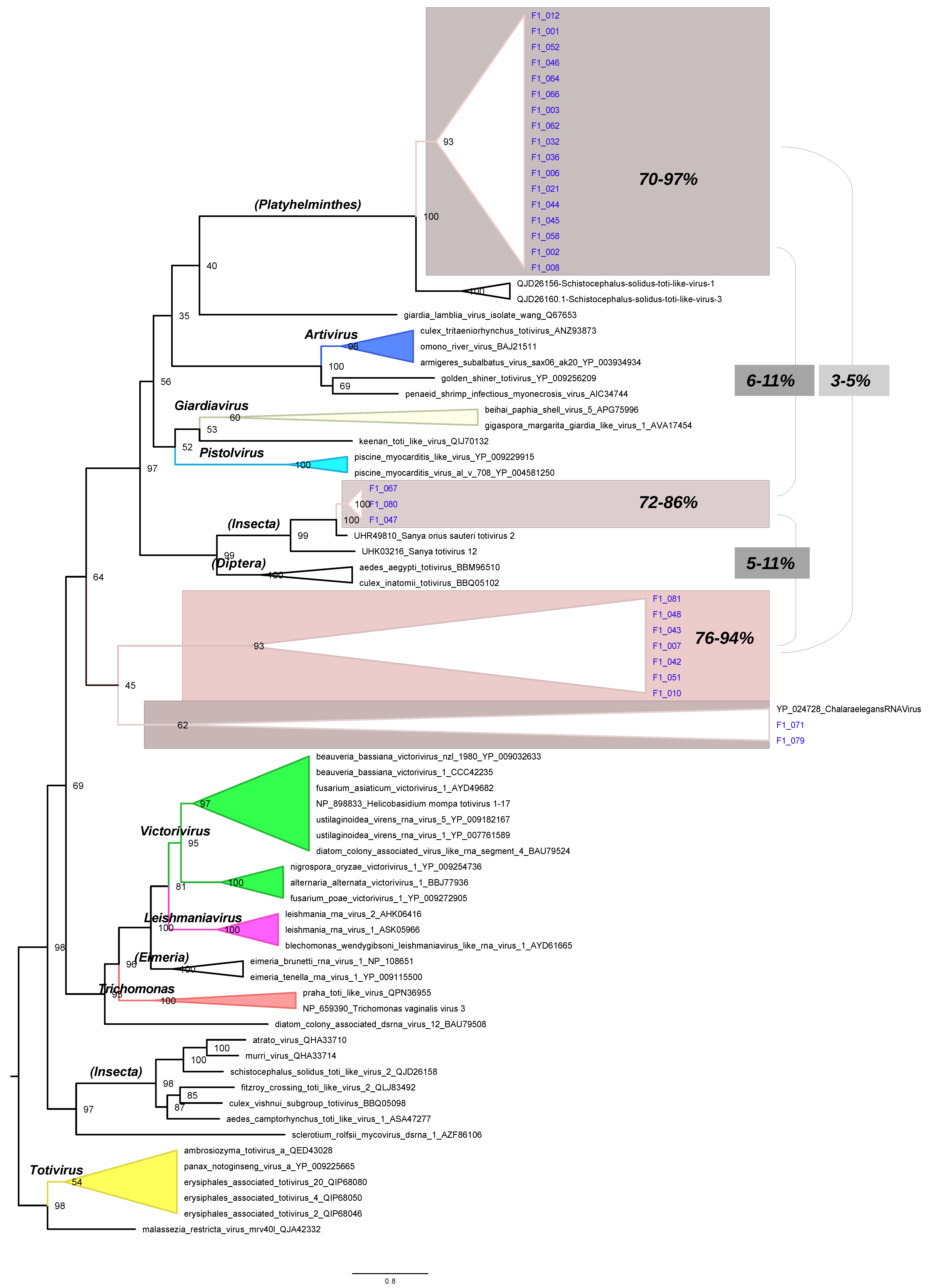
3.4. Phylogenetic Tree of RdRpol of Totiviruses
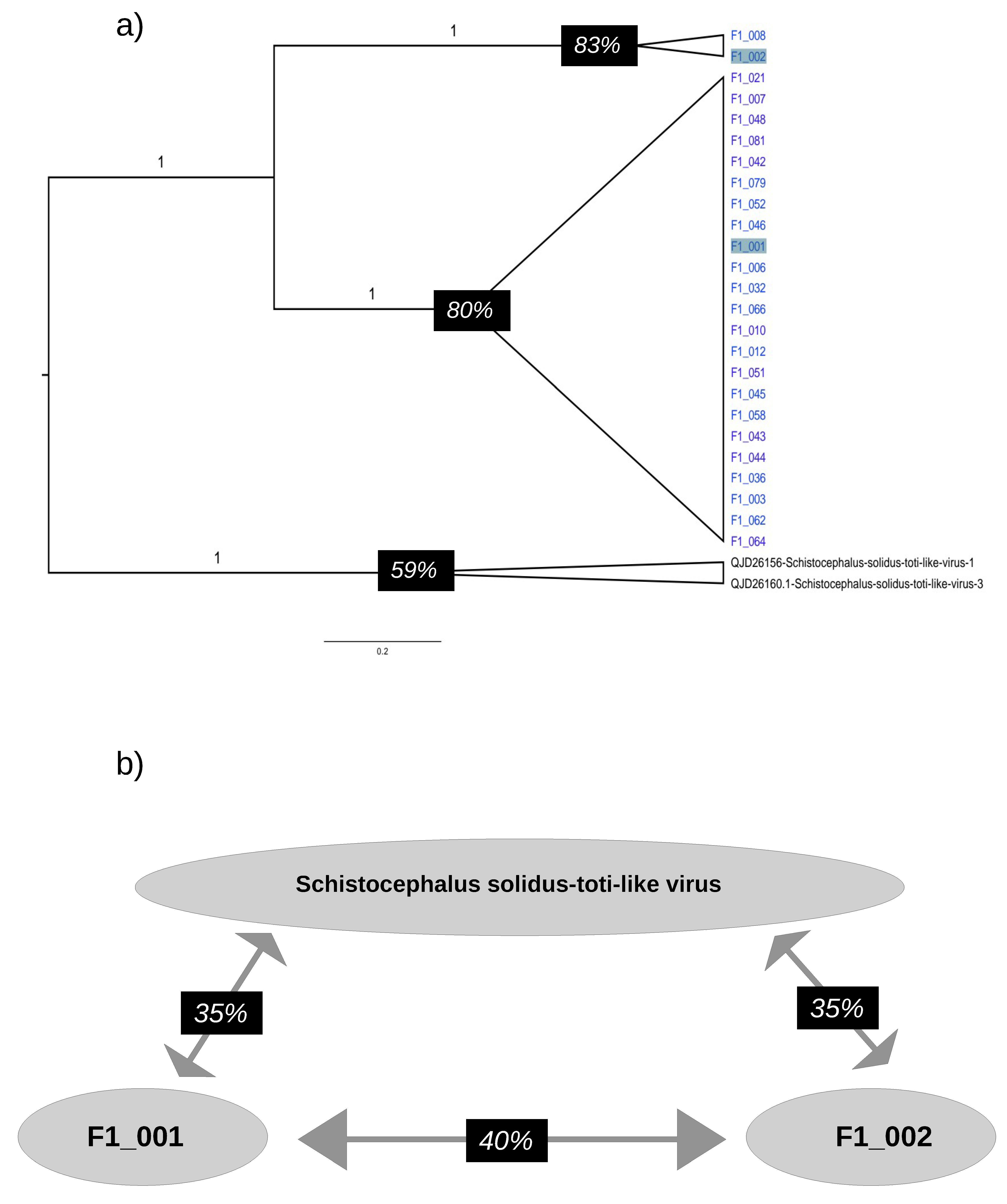
3.5. RdRpol Amino Acid Identity
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brook, C.E.; Dobson, A.P. Bats as ‘Special’ Reservoirs for Emerging Zoonotic Pathogens. Trends Microbiol. 2015, 23, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Moratelli, R.; Calisher, C.H. Bats and Zoonotic Viruses: Can We Confidently Link Bats with Emerging Deadly Viruses? Mem. Inst. Oswaldo Cruz 2015, 110, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Weekley, C.C.; Daszak, P. Are Bats Really “Special” as Viral Reservoirs? What We Know and Need to Know. In Bats and Viruses; Wang, L., Cowled, C., Eds.; Wiley: Hoboken, NJ, USA, 2015; pp. 281–294. ISBN 9781118818732. [Google Scholar]
- Castelo-Branco, D.S.C.M.; Nobre, J.A.; Souza, P.R.H.; Diógenes, E.M.; Guedes, G.M.M.; Mesquita, F.P.; Souza, P.F.N.; Rocha, M.F.G.; Sidrim, J.J.C.; Cordeiro, R.A.; et al. Role of Brazilian Bats in the Epidemiological Cycle of Potentially Zoonotic Pathogens. Microb. Pathog. 2023, 177, 106032. [Google Scholar] [CrossRef] [PubMed]
- Poel, W.H.M.V.D.; Lina, P.H.C.; Kramps, J.A. Public Health Awareness of Emerging Zoonotic Viruses of Bats: A European Perspective. Vector-Borne Zoonotic Dis. 2006, 6, 315–324. [Google Scholar] [CrossRef]
- Van Brussel, K.; Holmes, E.C. Zoonotic Disease and Virome Diversity in Bats. Curr. Opin. Virol. 2022, 52, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.M.; Hora, A.S.; Scheffer, K.C.; Fahl, W.O.; Iamamoto, K.; Mori, E.; Brandão, P.E. Alphacoronavirus in Urban Molossidae and Phyllostomidae Bats, Brazil. Virol. J. 2016, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Bittar, C.; Machado, R.R.G.; Comelis, M.T.; Bueno, L.M.; Beguelini, M.R.; Morielle-Versute, E.; Nogueira, M.L.; Rahal, P. Alphacoronavirus Detection in Lungs, Liver, and Intestines of Bats from Brazil. Microb. Ecol. 2020, 79, 203–212. [Google Scholar] [CrossRef]
- Campos, A.C.A.; Góes, L.G.B.; Moreira-Soto, A.; De Carvalho, C.; Ambar, G.; Sander, A.-L.; Fischer, C.; Ruckert Da Rosa, A.; Cardoso De Oliveira, D.; Kataoka, A.P.G.; et al. Bat Influenza A (HL18NL11) Virus in Fruit Bats, Brazil. Emerg. Infect. Dis. 2019, 25, 333–337. [Google Scholar] [CrossRef]
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; De Vries, M.; Drexler, J.F.; Oppong, S.K.; Müller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two Novel Parvoviruses in Frugivorous New and Old World Bats. PLoS ONE 2011, 6, e29140. [Google Scholar] [CrossRef]
- Cibulski, S.P.; Teixeira, T.F.; De Sales Lima, F.E.; Do Santos, H.F.; Franco, A.C.; Roehe, P.M. A Novel Anelloviridae Species Detected in Tadarida Brasiliensis Bats: First Sequence of a Chiropteran Anellovirus. Genome Announc. 2014, 2, e01028-14. [Google Scholar] [CrossRef]
- Cibulski, S.P.; De Sales Lima, F.E.; Teixeira, T.F.; Varela, A.P.M.; Scheffer, C.M.; Mayer, F.Q.; Witt, A.A.; Roehe, P.M. Detection of Multiple Viruses in Oropharyngeal Samples from Brazilian Free-Tailed Bats (Tadarida Brasiliensis) Using Viral Metagenomics. Arch. Virol. 2021, 166, 207–212. [Google Scholar] [CrossRef]
- Corman, V.M.; Rasche, A.; Diallo, T.D.; Cottontail, V.M.; Stöcker, A.; Souza, B.F.D.C.D.; Corrêa, J.I.; Carneiro, A.J.B.; Franke, C.R.; Nagy, M.; et al. Highly Diversified Coronaviruses in Neotropical Bats. J. Gen. Virol. 2013, 94, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, J.; Thomazelli, L.M.; Henriques, D.A.; Lautenschalager, D.; Ometto, T.; Dutra, L.M.; Aires, C.C.; Favorito, S.; Durigon, E.L. Detection of Hantavirus in Bats from Remaining Rain Forest in São Paulo, Brazil. BMC Res. Notes 2012, 5, 690. [Google Scholar] [CrossRef] [PubMed]
- Razafindratsimandresy, R.; Jeanmaire, E.M.; Counor, D.; Vasconcelos, P.F.; Sall, A.A.; Reynes, J.-M. Partial Molecular Characterization of Alphaherpesviruses Isolated from Tropical Bats. J. Gen. Virol. 2009, 90, 44–47. [Google Scholar] [CrossRef]
- Lima, F.E.D.S.; Cibulski, S.P.; Dos Santos, H.F.; Teixeira, T.F.; Varela, A.P.M.; Roehe, P.M.; Delwart, E.; Franco, A.C. Genomic Characterization of Novel Circular ssDNA Viruses from Insectivorous Bats in Southern Brazil. PLoS ONE 2015, 10, e0118070. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats Host Major Mammalian Paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Threlfall, C.G. Urbanisation and Its Effects on Bats—A Global Meta-Analysis; Voigt, C.C., Kingston, T., Eds.; Springer: Cham, Switzerland, 2016; pp. 13–33. [Google Scholar]
- Piló, L.B.; Calux, A.; Scherer, R.; Bernard, E. Bats as Ecosystem Engineers in Iron Ore Caves in the Carajás National Forest, Brazilian Amazonia. PLoS ONE 2023, 18, e0267870. [Google Scholar] [CrossRef]
- Wallau, G.L.; Barbier, E.; Tomazatos, A.; Schmidt-Chanasit, J.; Bernard, E. The Virome of Bats Inhabiting Brazilian Biomes: Knowledge Gaps and Biases towards Zoonotic Viruses. Microbiol. Spectr. 2023, 11, e04077-22. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the Bat Virome Catalog to Better Understand the Ecological Diversity of Bat Viruses and the Bat Origin of Emerging Infectious Diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef]
- Olivier, T.; Thébault, E.; Elias, M.; Fontaine, B.; Fontaine, C. Urbanization and Agricultural Intensification Destabilize Animal Communities Differently than Diversity Loss. Nat. Commun. 2020, 11, 2686. [Google Scholar] [CrossRef]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological Dynamics of Emerging Bat Virus Spillover. Proc. R. Soc. B 2015, 282, 20142124. [Google Scholar] [CrossRef] [PubMed]
- Luis, A.D.; O’Shea, T.J.; Hayman, D.T.S.; Wood, J.L.N.; Cunningham, A.A.; Gilbert, A.T.; Mills, J.N.; Webb, C.T. Network Analysis of Host–Virus Communities in Bats and Rodents Reveals Determinants of Cross-species Transmission. Ecol. Lett. 2015, 18, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Armero, A.; Li, R.; Bienes, K.M.; Chen, X.; Li, J.; Xu, S.; Chen, Y.; Hughes, A.C.; Berthet, N.; Wong, G. Myotis Fimbriatus Virome, a Window to Virus Diversity and Evolution in the Genus Myotis. Viruses 2022, 14, 1899. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic Analysis of the Viromes of Three North American Bat Species: Viral Diversity among Different Bat Species That Share a Common Habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhu, C.; Wang, Y.; Ai, L.; Yang, L.; Ye, F.; Ding, C.; Chen, J.; He, B.; Zhu, J.; et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bats from Southeast China. Sci. Rep. 2017, 7, 10917. [Google Scholar] [CrossRef]
- Bueno, L.M.; Rizotto, L.S.; Viana, A.D.O.; Silva, L.M.N.; De Moraes, M.V.D.S.; Benassi, J.C.; Scagion, G.P.; Dorlass, E.G.; Lopes, B.L.T.; Cunha, I.N.; et al. High Genetic Diversity of Alphacoronaviruses in Bat Species (Mammalia: Chiroptera) from the Atlantic Forest in Brazil. Transbound. Emerg. Dis. 2022, 69, e2863–e2875. [Google Scholar] [CrossRef]
- Ramos, E.D.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.D.S.; Villanova, F.; Pandey, R.P.; Araújo, E.L.L.; Deng, X.; Delwart, E.; et al. Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region. Viruses 2023, 15, 606. [Google Scholar] [CrossRef]
- Salmier, A.; Tirera, S.; De Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome Analysis of Two Sympatric Bat Species (Desmodus Rotundus and Molossus Molossus) in French Guiana. PLoS ONE 2017, 12, e0186943. [Google Scholar] [CrossRef]
- Kohl, C.; Brinkmann, A.; Radonić, A.; Dabrowski, P.W.; Mühldorfer, K.; Nitsche, A.; Wibbelt, G.; Kurth, A. The Virome of German Bats: Comparing Virus Discovery Approaches. Sci. Rep. 2021, 11, 7430. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Ge, X.; Yuan, J.; Shi, Z. A Novel Totivirus-like Virus Isolated from Bat Guano. Arch. Virol. 2012, 157, 1093–1099. [Google Scholar] [CrossRef]
- Call, L.; Nayfach, S.; Kyrpides, N.C. Illuminating the Virosphere Through Global Metagenomics. Annu. Rev. Biomed. Data Sci. 2021, 4, 369–391. [Google Scholar] [CrossRef]
- Totiviridae. ICTV. Available online: https://ictv.global/report_9th/dsRNA/Totiviridae (accessed on 23 November 2023).
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; Diclaro, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles Gambiae Mosquitoes Harbor a Taxonomically Diverse Virome Including New Insect-Specific Flaviviruses, Mononegaviruses, and Totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, X.; Wu, W.; Tan, G.; Fang, S.; Zhang, S.; Li, F. Identification and Molecular Characterization of Panax Notoginseng Virus A, Which May Represent an Undescribed Novel Species of the Genus Totivirus, Family Totiviridae. Arch. Virol. 2016, 161, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, M.E.; MacDiarmid, R.M. A Novel Totivirus Naturally Occurring in Two Different Fungal Genera. Front. Microbiol. 2019, 10, 2318. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Du, J.; Wu, Z.; Zhang, W.; Fu, S.; Song, J.; Wang, Q.; He, Y.; Lei, W.; Xu, S.; et al. Identification and Genetic Analysis of a Totivirus Isolated from the Culex Tritaeniorhynchus in Northern China. Arch. Microbiol. 2020, 202, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Løvoll, M.; Wiik-Nielsen, J.; Grove, S.; Wiik-Nielsen, C.R.; Kristoffersen, A.B.; Faller, R.; Poppe, T.; Jung, J.; Pedamallu, C.S.; Nederbragt, A.J.; et al. A Novel Totivirus and Piscine Reovirus (PRV) in Atlantic Salmon (Salmo Salar) with Cardiomyopathy Syndrome (CMS). Virol. J. 2010, 7, 309. [Google Scholar] [CrossRef]
- Okamoto, K.; Miyazaki, N.; Larsson, D.S.D.; Kobayashi, D.; Svenda, M.; Mühlig, K.; Maia, F.R.N.C.; Gunn, L.H.; Isawa, H.; Kobayashi, M.; et al. The Infectious Particle of Insect-Borne Totivirus-like Omono River Virus Has Raised Ridges and Lacks Fibre Complexes. Sci. Rep. 2016, 6, 33170. [Google Scholar] [CrossRef]
- Park, Y.; James, D.; Punja, Z.K. Co-Infection by Two Distinct Totivirus-like Double-Stranded RNA Elements in Chalara Elegans (Thielaviopsis Basicola). Virus Res. 2005, 109, 71–85. [Google Scholar] [CrossRef]
- Qu, J.; Shi, N.; Yang, G.; Huang, B. Molecular Characterization of a Novel Totivirus Infecting the Basal Fungus Conidiobolus Heterosporus. Arch. Virol. 2021, 166, 1801–1804. [Google Scholar] [CrossRef]
- Mor, S.K.; Phelps, N.B.D. Molecular Detection of a Novel Totivirus from Golden Shiner (Notemigonus Crysoleucas) Baitfish in the USA. Arch. Virol. 2016, 161, 2227–2234. [Google Scholar] [CrossRef]
- Janssen, M.E.W.; Takagi, Y.; Parent, K.N.; Cardone, G.; Nibert, M.L.; Baker, T.S. Three-Dimensional Structure of a Protozoal Double-Stranded RNA Virus That Infects the Enteric Pathogen Giardia Lamblia. J. Virol. 2015, 89, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Procházková, M.; Füzik, T.; Grybchuk, D.; Falginella, F.L.; Podešvová, L.; Yurchenko, V.; Vácha, R.; Plevka, P. Capsid Structure of Leishmania RNA Virus 1. J. Virol. 2021, 95, e01957-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.J.; Zhong, J.; Shang, H.H.; Pan, X.T.; Zhu, H.J.; Da Gao, B. The Complete Nucleotide Sequence and Genomic Organization of a Novel Victorivirus with Two Non-Overlapping ORFs, Identified in the Plant-Pathogenic Fungus Phomopsis Vexans. Arch. Virol. 2015, 160, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Castón, J.R.; Luque, D.; Trus, B.L.; Rivas, G.; Alfonso, C.; González, J.M.; Carrascosa, J.L.; Annamalai, P.; Ghabrial, S.A. Three-Dimensional Structure and Stoichiometry of Helmintosporium victoriae 190S Totivirus. Virology 2006, 347, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Ghabrial, S.A.; Fichorova, R.N.; Nibert, M.L. Trichomonasvirus: A New Genus of Protozoan Viruses in the Family Totiviridae. Arch. Virol. 2011, 156, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Fraga, J.; Rojas, L.; Sariego, I.; Fernández-Calienes, A. Genetic Characterization of Three Cuban Trichomonas Vaginalis Virus. Phylogeny of Totiviridae Family. Infect. Genet. Evol. 2012, 12, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; De Matos Filipe, D.; Okamoto, K. A Full-Length Infectious cDNA Clone of a dsRNA Totivirus-like Virus. Virology 2022, 576, 127–133. [Google Scholar] [CrossRef]
- Burrows, J.T.A.; Depierreux, D.; Nibert, M.L.; Pearson, B.J. A Novel Taxon of Monosegmented Double-Stranded RNA Viruses Endemic to Triclad Flatworms. J. Virol. 2020, 94, e00623-20. [Google Scholar] [CrossRef]
- Sandlund, L.; Mor, S.K.; Singh, V.K.; Padhi, S.K.; Phelps, N.B.D.; Nylund, S.; Mikalsen, A.B. Comparative Molecular Characterization of Novel and Known Piscine Toti-Like Viruses. Viruses 2021, 13, 1063. [Google Scholar] [CrossRef]
- Zhao, M.; Xu, L.; Bowers, H.; Schott, E.J. Characterization of Two Novel Toti-Like Viruses Co-Infecting the Atlantic Blue Crab, Callinectes Sapidus, in Its Northern Range of the United States. Front. Microbiol. 2022, 13, 855750. [Google Scholar] [CrossRef]
- Zhai, Y.; Attoui, H.; Mohd Jaafar, F.; Wang, H.-Q.; Cao, Y.-X.; Fan, S.-P.; Sun, Y.-x.; Liu, L.-D.; Mertens, P.P.C.; Meng, W.-S.; et al. Isolation and Full-Length Sequence Analysis of Armigeres Subalbatus Totivirus, the First Totivirus Isolate from Mosquitoes Representing a Proposed Novel Genus (Artivirus) of the Family Totiviridae. J. Gen. Virol. 2010, 91, 2836–2845. [Google Scholar] [CrossRef] [PubMed]
- Sassa, Y.; Ono, S.; Takata, M.; Koyama, S.; Furuya, T.; Satoh, T.; Ohmatsu, T.; Nagai, M.; Sakai, C.; Mizutani, T.; et al. Identification, Characterization and Full-Length Sequence Analysis of a Novel dsRNA Virus Isolated from the Arboreal Ant Camponotus Yamaokai. J. Gen. Virol. 2015, 96, 1930–1937. [Google Scholar] [CrossRef]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An Ensemble Strategy That Significantly Improves de Novo Assembly of Microbial Genomes from Metagenomic Next-Generation Sequencing Data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. The UGENE team Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary Trees from DNA Sequences: A Maximum Likelihood Approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Rosario, K.; Lucas, P.; Dheilly, N.M. Characterization of Viruses in a Tapeworm: Phylogenetic Position, Vertical Transmission, and Transmission to the Parasitized Host. ISME J. 2020, 14, 1755–1767. [Google Scholar] [CrossRef]
- Lima, F.E.D.S.; Cibulski, S.P.; Elesbao, F.; Carnieli, P., Jr.; Batista, H.B.D.C.R.; Roehe, P.M.; Franco, A.C. First Detection of Adenovirus in the Vampire Bat (Desmodus Rotundus) in Brazil. Virus Genes 2013, 47, 378–381. [Google Scholar] [CrossRef]
- Góes, L.G.B.; Campos, A.C.D.A.; Carvalho, C.D.; Ambar, G.; Queiroz, L.H.; Cruz-Neto, A.P.; Munir, M.; Durigon, E.L. Genetic Diversity of Bats Coronaviruses in the Atlantic Forest Hotspot Biome, Brazil. Infect. Genet. Evol. 2016, 44, 510–513. [Google Scholar] [CrossRef]
- Aguilar Pierlé, S.; Zamora, G.; Ossa, G.; Gaggero, A.; Barriga, G.P. The Myotis Chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly. Viruses 2022, 14, 202. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couto, R.d.S.; Ramos, E.d.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.d.S.; Villanova, F.; Pandey, R.P.; Deng, X.; Delwart, E.; et al. Metagenomic of Liver Tissue Identified at Least Two Genera of Totivirus-like Viruses in Molossus molossus Bats. Microorganisms 2024, 12, 206. https://doi.org/10.3390/microorganisms12010206
Couto RdS, Ramos EdSF, Abreu WU, Rodrigues LRR, Marinho LF, Morais VdS, Villanova F, Pandey RP, Deng X, Delwart E, et al. Metagenomic of Liver Tissue Identified at Least Two Genera of Totivirus-like Viruses in Molossus molossus Bats. Microorganisms. 2024; 12(1):206. https://doi.org/10.3390/microorganisms12010206
Chicago/Turabian StyleCouto, Roseane da Silva, Endrya do Socorro Foro Ramos, Wandercleyson Uchôa Abreu, Luis Reginaldo Ribeiro Rodrigues, Luis Fernando Marinho, Vanessa dos Santos Morais, Fabiola Villanova, Ramendra Pati Pandey, Xutao Deng, Eric Delwart, and et al. 2024. "Metagenomic of Liver Tissue Identified at Least Two Genera of Totivirus-like Viruses in Molossus molossus Bats" Microorganisms 12, no. 1: 206. https://doi.org/10.3390/microorganisms12010206
APA StyleCouto, R. d. S., Ramos, E. d. S. F., Abreu, W. U., Rodrigues, L. R. R., Marinho, L. F., Morais, V. d. S., Villanova, F., Pandey, R. P., Deng, X., Delwart, E., Costa, A. C. d., & Leal, E. (2024). Metagenomic of Liver Tissue Identified at Least Two Genera of Totivirus-like Viruses in Molossus molossus Bats. Microorganisms, 12(1), 206. https://doi.org/10.3390/microorganisms12010206