
An Analysis of Combined Molecular Weight and Hydrophobicity Similarity between the Amino Acid Sequences of Spike Protein Receptor Binding Domains of Betacoronaviruses and Functionally Similar Sequences from Other Virus Families
Abstract
:1. Introduction
2. Materials and Methods
2.1. Waveform-Based Hierarchical Clustering
2.2. Pairwise Global Alignment
2.3. In Silico Structural Design
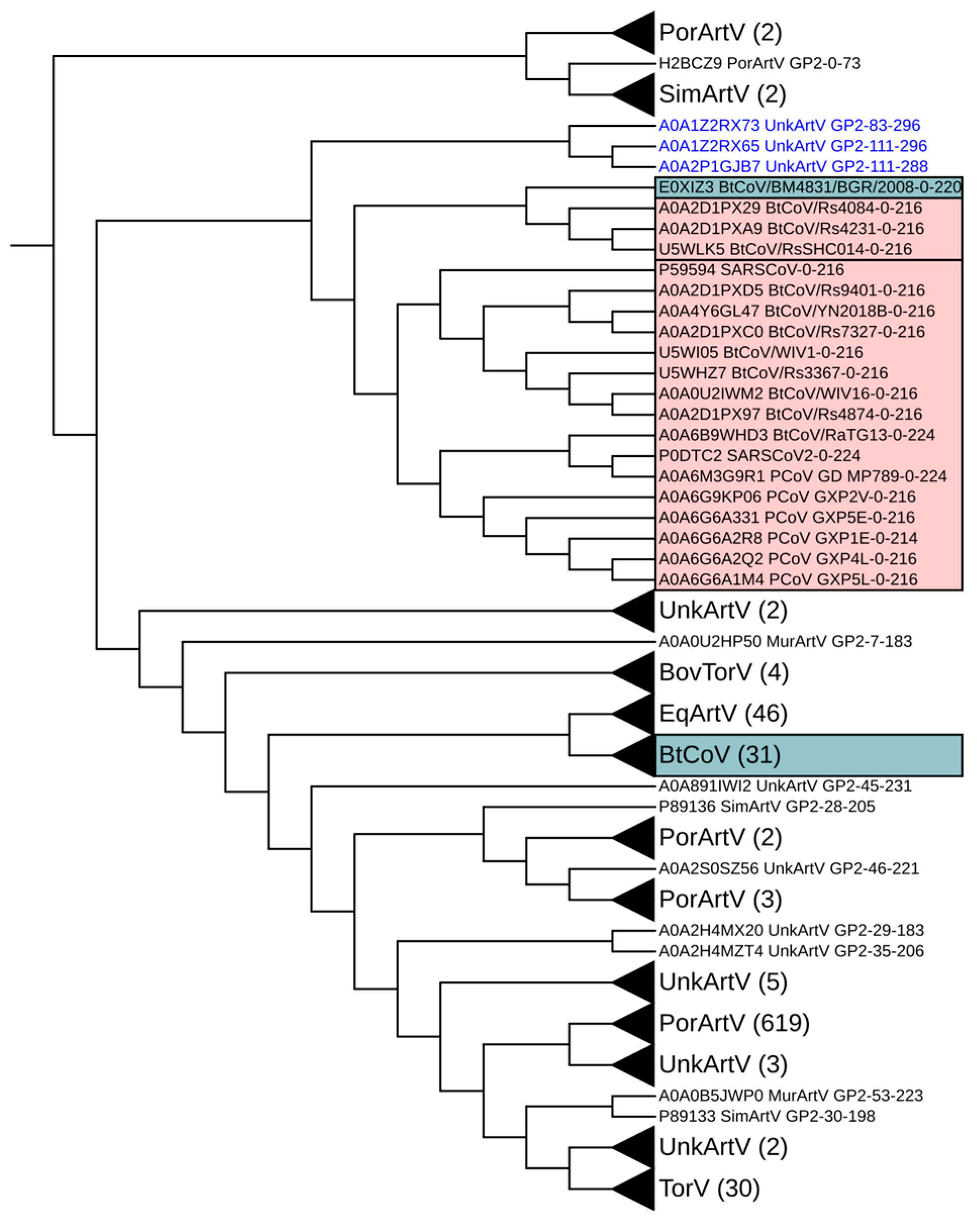
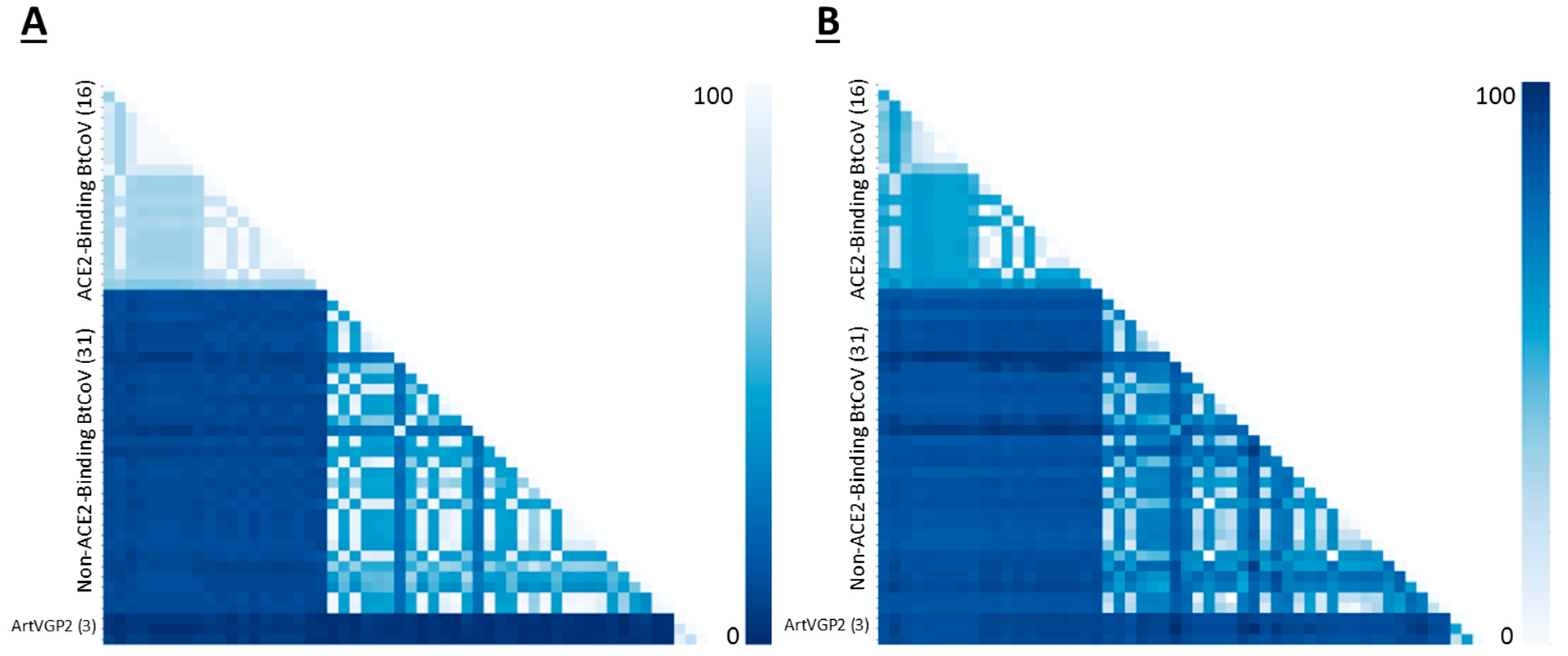
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization WHO | Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Available online: https://www.who.int/emergencies/mers-cov/en/ (accessed on 16 September 2020).
- World Health Organization Severe Acute Respiratory Syndrome (SARS). Available online: https://www.who.int/health-topics/severe-acute-respiratory-syndrome#tab=tab_1 (accessed on 17 September 2020).
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Al Asmari, M.; Islam, A.; Kapoor, A.; et al. Middle East Respiratory Syndrome Coronavirus in Bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.M.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Wells, H.L.; Letko, M.; Lasso, G.; Ssebide, B.; Nziza, J.; Byarugaba, D.K.; Navarrete-Macias, I.; Liang, E.; Cranfield, M.; Han, B.A.; et al. The Evolutionary History of ACE2 Usage within the Coronavirus Subgenus Sarbecovirus. Virus Evol. 2021, 7, veab007. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the Origin and Continuing Evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef]
- Lam, T.T.Y.; Jia, N.; Zhang, Y.W.; Shum, M.H.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-Related Coronaviruses in Malayan Pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Malaiyan, J.; Arumugam, S.; Mohan, K.; Gomathi Radhakrishnan, G. An Update on the Origin of SARS-CoV-2: Despite Closest Identity, Bat (RaTG13) and Pangolin Derived Coronaviruses Varied in the Critical Binding Site and O-Linked Glycan Residues. J. Med. Virol. 2021, 93, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Dixson, J.D.; Azad, R.K. A Novel Predictor of ACE2-Binding Ability among Betacoronaviruses. Evol. Med. Public Health 2021, 9, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Nonhomologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2022, 39, msab292. [Google Scholar] [CrossRef]
- Dixson, J.D.; Azad, R.K. Physicochemical Evaluation of Remote Homology in the Twilight Zone. Proteins Struct. Funct. Bioinform. 2024. [Google Scholar] [CrossRef]
- Storz, J.F. Causes of Molecular Convergence and Parallelism in Protein Evolution. Nat. Rev. Genet. 2016, 17, 239–250. [Google Scholar] [CrossRef]
- World Health Organization WHO Coronavirus Disease (COVID-19) Dashboard | WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 October 2024).
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: A Comparative Overview. Le Infez. Med. 2020, 28, 174–184. [Google Scholar]
- Locatelli, I.; Trächsel, B.; Rousson, V. Estimating the Basic Reproduction Number for COVID-19 in Western Europe. PLoS ONE 2021, 16, e0248731. [Google Scholar] [CrossRef]
- Rahman, M.M.; Masum, M.H.U.; Wajed, S.; Talukder, A. A Comprehensive Review on COVID-19 Vaccines: Development, Effectiveness, Adverse Effects, Distribution and Challenges. Virus Dis. 2022, 33, 1–22. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; Filip, I.; Rabadan, R. Global Patterns of Recombination across Human Viruses. Mol. Biol. Evol. 2021, 38, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Galindo, J.Á.; Filip, I.; Chowdhury, R.; Maranas, C.D.; Sorger, P.K.; AlQuraishi, M.; Rabadan, R. Recombination and Lineage-Specific Mutations Linked to the Emergence of SARS-CoV-2. Genome Med. 2021, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 9781493924387. [Google Scholar]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between Nonsegmented RNA Genomes of Murine Coronaviruses. J. Virol. 1985, 56, 449. [Google Scholar] [CrossRef]
- Keck, J.G.; Makino, S.; Soe, L.H.; Fleming, J.O.; Stohlman, S.A.; Lai, M.M.C. RNA Recombination of Coronavirus. Adv. Exp. Med. Biol. 1987, 218, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Giorg, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through Recombination and Strong Purifying Selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Zhu, Z.; Meng, K.; Meng, G. Genomic Recombination Events May Reveal the Evolution of Coronavirus and the Origin of SARS-CoV-2. Sci. Rep. 2020, 10, 21617. [Google Scholar] [CrossRef]
- Desbiez, C.; Joannon, B.; Wipf-Scheibel, C.; Chandeysson, C.; Lecoq, H. Recombination in Natural Populations of Watermelon Mosaic Virus: New Agronomic Threat or Damp Squib? J. Gen. Virol. 2011, 92, 1939–1948. [Google Scholar] [CrossRef]
- Miras, M.; Sempere, R.N.; Kraft, J.J.; Miller, W.A.; Aranda, M.A.; Truniger, V. Interfamilial Recombination between Viruses Led to Acquisition of a Novel Translation-Enhancing RNA Element That Allows Resistance Breaking. New Phytol. 2014, 202, 233–246. [Google Scholar] [CrossRef]
- Chapter 25—Arteriviridae and Roniviridae. In Fenner’s Veterinary Virology; MacLachlan, N.J.; Dubovi, E.J. (Eds.) Academic Press (Elsevier): London, UK, 2017; pp. 463–476. ISBN 978-0-12-800946-8. [Google Scholar]
- Veit, M.; Matczuk, A.K.; Sinhadri, B.C.; Krause, E.; Thaa, B. Membrane Proteins of Arterivirus Particles: Structure, Topology, Processing and Function. Virus Res. 2014, 194, 16–36. [Google Scholar] [CrossRef]
- Jiang, R.-D.; Li, B.; Liu, X.-L.; Liu, M.-Q.; Chen, J.; Luo, D.-S.; Hu, B.-J.; Zhang, W.; Li, S.-Y.; Yang, X.-L.; et al. Bat Mammalian Orthoreoviruses Cause Severe Pneumonia in Mice. Virology 2020, 551, 84–92. [Google Scholar] [CrossRef]
- Simsek, C.; Corman, V.M.; Everling, H.U.; Lukashev, A.N.; Rasche, A.; Maganga, G.D.; Binger, T.; Jansen, D.; Beller, L.; Deboutte, W.; et al. At Least Seven Distinct Rotavirus Genotype Constellations in Bats with Evidence of Reassortment and Zoonotic Transmissions. MBio 2021, 12, e02755-20. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.R.; Gaby, J.E. Dynamic Waveform Matching. Inf. Sci. 1983, 31, 221–242. [Google Scholar] [CrossRef]
- Sokal, R.R.; Michener, C.D. A Statistical Method for Evaluating Relationships. Univ. Kansas Sci. Bull. 1958, 38, 1409–1448. [Google Scholar]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R Package for Visualizing, Adjusting, and Comparing Trees of Hierarchical Clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Smith, M.R. TreeDist: Distances between Phylogenetic Trees. R Package Version 2.0.3. Available online: https://cran.r-project.org/web/packages/%0ATreeTools/index.html (accessed on 1 October 2024).
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 127–147. [Google Scholar] [CrossRef]
- Fan, H.H.; Wang, L.Q.; Liu, W.L.; An, X.P.; Liu, Z.D.; He, X.Q.; Song, L.H.; Tong, Y.G. Repurposing of Clinically Approved Drugs for Treatment of Coronavirus Disease 2019 in a 2019-Novel Coronavirus-Related Coronavirus Model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Calder, L.J.; Borg, A.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Structure and Binding Properties of Pangolin-CoV Spike Glycoprotein Inform the Evolution of SARS-CoV-2. Nat. Commun. 2021, 12, 837. [Google Scholar] [CrossRef]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and Characterization of a Bat SARS-like Coronavirus That Uses the ACE2 Receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. A SARS-like Cluster of Circulating Bat Coronaviruses Shows Potential for Human Emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for Structure Building and Analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Rost, B. Twilight Zone of Protein Sequence Alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Jain, A.; Perisa, D.; Fliedner, F.; von Haeseler, A.; Ebersberger, I. The Evolutionary Traceability of a Protein. Genome Biol. Evol. 2019, 11, 531–545. [Google Scholar] [CrossRef]
- Gascuel, O.; Steel, M. Neighbor-Joining Revealed. Mol. Biol. Evol. 2006, 23, 1997–2000. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, J.S.; Slotboom, D.-J. Hydropathy Profile Alignment: A Tool to Search for Structural Homologues of Membrane Proteins. FEMS Microbiol. Rev. 1998, 22, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D.; Smith, N.G.C. The Evolution of Concerted Evolution. Proc. R. Soc. Biol. Sci. 1998, 265, 121–127. [Google Scholar] [CrossRef]
- Kumar, S.; Filipski, A.J.; Battistuzzi, F.U.; Kosakovsky Pond, S.L.; Tamura, K. Statistics and Truth in Phylogenomics. Mol. Biol. Evol. 2012, 29, 457–472. [Google Scholar] [CrossRef]
- Jia, K.; Jernigan, R.L. New Amino Acid Substitution Matrix Brings Sequence Alignments into Agreement with Structure Matches. Proteins 2021, 89, 671–682. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Value | AA | Value | AA | Value |
|---|---|---|---|---|---|
| G | 0.3064 | Q | −6.8806 | Y | 42.9375 |
| A | 15.6497 | E | 5.547 | H | −22.8297 |
| L | 60.7668 | S | −2.3381 | R | −11.7423 |
| M | 52.1362 | P | −23.9905 | N | −17.1576 |
| F | 76.6645 | V | 40.4569 | D | −33.9918 |
| W | 97.0000 | I | 60.1592 | T | 7.0585 |
| K | −15.8315 | C | 27.1418 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixson, J.D.; Vumma, L.; Azad, R.K. An Analysis of Combined Molecular Weight and Hydrophobicity Similarity between the Amino Acid Sequences of Spike Protein Receptor Binding Domains of Betacoronaviruses and Functionally Similar Sequences from Other Virus Families. Microorganisms 2024, 12, 2021. https://doi.org/10.3390/microorganisms12102021
Dixson JD, Vumma L, Azad RK. An Analysis of Combined Molecular Weight and Hydrophobicity Similarity between the Amino Acid Sequences of Spike Protein Receptor Binding Domains of Betacoronaviruses and Functionally Similar Sequences from Other Virus Families. Microorganisms. 2024; 12(10):2021. https://doi.org/10.3390/microorganisms12102021
Chicago/Turabian StyleDixson, Jamie D., Lavanya Vumma, and Rajeev K. Azad. 2024. "An Analysis of Combined Molecular Weight and Hydrophobicity Similarity between the Amino Acid Sequences of Spike Protein Receptor Binding Domains of Betacoronaviruses and Functionally Similar Sequences from Other Virus Families" Microorganisms 12, no. 10: 2021. https://doi.org/10.3390/microorganisms12102021