
SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins
 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved Response to Emergent Viral Outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Euro Surveill. Bull. Eur. Sur Mal. Transm. Eur. Commun. Dis. Bull. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; VanInsberghe, D.; Koelle, K. Insights from SARS-CoV-2 Sequences. Science 2021, 371, 466–467. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Ba, A.A.; Coppée, R.; Dieng, A.; Manneh, J.; Fall, M.; Gueye, K.; Sene, P.Y.; Ndiour, S.; Samaté, D.; Manga, P.; et al. Genomic Epidemiology of SARS-CoV-2 in Senegal in 2020–2021. J. Infect. Dev. Ctries. 2024, 18, 851–861. [Google Scholar] [CrossRef]
- Machado, L.C.; Dezordi, F.Z.; de Lima, G.B.; de Lima, R.E.; Silva, L.C.A.; Pereira, L.d.M.; da Silva, A.F.; Silva Neto, A.M.d.; Oliveira, A.L.S.d.; Armstrong, A.d.C.; et al. Spatiotemporal Transmission of SARS-CoV-2 Lineages during 2020-2021 in Pernambuco-Brazil. Microbiol. Spectr. 2024, 12, e0421823. [Google Scholar] [CrossRef]
- Khairnar, K.; Tomar, S.S. COVID-19 Genome Surveillance: A Geographical Landscape and Mutational Mapping of SARS-CoV-2 Variants in Central India over Two Years. Virus Res. 2024, 344, 199365. [Google Scholar] [CrossRef]
- Lagare, A.; Faye, M.; Issa, M.; Hamidou, O.; Bienvenu, B.; Mohamed, A.; Aoula, B.; Moumouni, K.; Hassane, F.; Otto, Y.A.; et al. First Identification of the SARS-COV-2/XBB.1.5 Sublineage among Indigenous COVID-19 Cases through the Influenza Sentinel Surveillance System in Niger. Heliyon 2023, 9, e20916. [Google Scholar] [CrossRef]
- Walensky, R.P.; Walke, H.T.; Fauci, A.S. SARS-CoV-2 Variants of Concern in the United States-Challenges and Opportunities. JAMA 2021, 325, 1037–1038. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next Generation Sequencing of SARS-CoV-2 Genomes: Challenges, Applications and Opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Charron, J.; Stenger, C.L.; Painter, J.; Steward, H.; Cook, T.W.; Faber, W.; Frisch, A.; Lind, E.; Bauss, J.; et al. SARS-CoV-2 (COVID-19) Structural and Evolutionary Dynamicome: Insights into Functional Evolution and Human Genomics. J. Biol. Chem. 2020, 295, 11742–11753. [Google Scholar] [CrossRef]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, T.; Sonnhammer, E.L.L. Kalign—An Accurate and Fast Multiple Sequence Alignment Algorithm. BMC Bioinform. 2005, 6, 298. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE team Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis Software for Microcomputers. Comput. Appl. Biosci. CABIOS 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The Worldwide Protein Data Bank (wwPDB): Ensuring a Single, Uniform Archive of PDB Data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef]
- Yan, L.; Yang, Y.; Li, M.; Zhang, Y.; Zheng, L.; Ge, J.; Huang, Y.C.; Liu, Z.; Wang, T.; Gao, S.; et al. Coupling of N7-Methyltransferase and 3′-5′ Exoribonuclease with SARS-CoV-2 Polymerase Reveals Mechanisms for Capping and Proofreading. Cell 2021, 184, 3474–3485.e11. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and Inhibition of the Papain-like Protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Brewitz, L.; Dumjahn, L.; Zhao, Y.; Owen, C.D.; Laidlaw, S.M.; Malla, T.R.; Nguyen, D.; Lukacik, P.; Salah, E.; Crawshaw, A.D.; et al. Alkyne Derivatives of SARS-CoV-2 Main Protease Inhibitors Including Nirmatrelvir Inhibit by Reacting Covalently with the Nucleophilic Cysteine. J. Med. Chem. 2023, 66, 2663–2680. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the Precision of Comparative Models with YASARA NOVA--a Self-Parameterizing Force Field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB Information Portal for Structural Genomics. Nucleic Acids Res. 2006, 34, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: The XBB.1.5 (‘Kraken’) Subvariant of Omicron SARS-CoV-2 and Its Rapid Global Spread. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2023, 29, e939580. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Coronavirus Variant XBB.1.5 Rises in the United States—Is It a Global Threat? Nature 2023, 613, 222–223. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Rahat, B.; Naqvi, B.; Rizvi, S.K.A. Impact of Covid-19 on Corporate Solvency and Possible Policy Responses in the EU. Q. Rev. Econ. Finance J. Midwest Econ. Assoc. 2023, 87, 181–190. [Google Scholar] [CrossRef]
- Alvarez, E.; Bielska, I.A.; Hopkins, S.; Belal, A.A.; Goldstein, D.M.; Slick, J.; Pavalagantharajah, S.; Wynfield, A.; Dakey, S.; Gedeon, M.-C.; et al. Limitations of COVID-19 Testing and Case Data for Evidence-Informed Health Policy and Practice. Health Res. Policy Syst. 2023, 21, 11. [Google Scholar] [CrossRef]
- Hartog, N.; Faber, W.; Frisch, A.; Bauss, J.; Bupp, C.P.; Rajasekaran, S.; Prokop, J.W. SARS-CoV-2 Infection: Molecular Mechanisms of Severe Outcomes to Suggest Therapeutics. Expert Rev. Proteomics 2021, 18, 105–118. [Google Scholar] [CrossRef]
- Sirpilla, O.; Bauss, J.; Gupta, R.; Underwood, A.; Qutob, D.; Freeland, T.; Bupp, C.; Carcillo, J.; Hartog, N.; Rajasekaran, S.; et al. SARS-CoV-2-Encoded Proteome and Human Genetics: From Interaction-Based to Ribosomal Biology Impact on Disease and Risk Processes. J. Proteome Res. 2020, 19, 4275–4290. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron Spike Trimer with ACE2 and an Anti-Omicron Antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Pastorio, C.; Zech, F.; Noettger, S.; Jung, C.; Jacob, T.; Sanderson, T.; Sparrer, K.M.J.; Kirchhoff, F. Determinants of Spike Infectivity, Processing, and Neutralization in SARS-CoV-2 Omicron Subvariants BA.1 and BA.2. Cell Host Microbe 2022, 30, 1255–1268.e5. [Google Scholar] [CrossRef]
- Verkhivker, G. Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2020, 21, 8268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Canena, D.; Sikora, M.; Klausberger, M.; Seferovic, H.; Mehdipour, A.R.; Hain, L.; Laurent, E.; Monteil, V.; Wirnsberger, G.; et al. Force-Tuned Avidity of Spike Variant-ACE2 Interactions Viewed on the Single-Molecule Level. Nat. Commun. 2022, 13, 7926. [Google Scholar] [CrossRef]
- Kakavandi, S.; Zare, I.; VaezJalali, M.; Dadashi, M.; Azarian, M.; Akbari, A.; Ramezani Farani, M.; Zalpoor, H.; Hajikhani, B. Structural and Non-Structural Proteins in SARS-CoV-2: Potential Aspects to COVID-19 Treatment or Prevention of Progression of Related Diseases. Cell Commun. Signal. CCS 2023, 21, 110. [Google Scholar] [CrossRef]
- Hynes, R.O. Cell Surface Proteins and Malignant Transformation. Biochim. Biophys. Acta 1976, 458, 73–107. [Google Scholar] [CrossRef]
- Choppin, P.W.; Scheid, A. The Role of Viral Glycoproteins in Adsorption, Penetration, and Pathogenicity of Viruses. Rev. Infect. Dis. 1980, 2, 40–61. [Google Scholar] [CrossRef]
- Hynes, R.O. Alteration of Cell-Surface Proteins by Viral Transformation and by Proteolysis. Proc. Natl. Acad. Sci. USA 1973, 70, 3170–3174. [Google Scholar] [CrossRef]
- Vossen, M.T.M.; Westerhout, E.M.; Söderberg-Nauclér, C.; Wiertz, E.J.H.J. Viral Immune Evasion: A Masterpiece of Evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef]
- Lynch, M. Mutation Pressure, Drift, and the Pace of Molecular Coevolution. Proc. Natl. Acad. Sci. USA 2023, 120, e2306741120. [Google Scholar] [CrossRef]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene Duplication as a Major Force in Evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and Evolution of Viruses of Eukaryotes: The Ultimate Modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef]
- Turner, B.G.; Summers, M.F. Structural Biology of HIV. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.Y.; Phatak, P.; Mukherjee, A. Cutting Edge Strategies for Screening of Novel Anti-HIV Drug Candidates against HIV Infection: A Concise Overview of Cell Based Assays. Heliyon 2023, 9, e16027. [Google Scholar] [CrossRef]
- Lu, I.-L.; Mahindroo, N.; Liang, P.-H.; Peng, Y.-H.; Kuo, C.-J.; Tsai, K.-C.; Hsieh, H.-P.; Chao, Y.-S.; Wu, S.-Y. Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef]
- Noble, C.G.; Shi, P.-Y. Structural Biology of Dengue Virus Enzymes: Towards Rational Design of Therapeutics. Antiviral Res. 2012, 96, 115–126. [Google Scholar] [CrossRef]
- Cox, B.D.; Stanton, R.A.; Schinazi, R.F. Predicting Zika Virus Structural Biology: Challenges and Opportunities for Intervention. Antivir. Chem. Chemother. 2015, 24, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.-M. Structural Biology of Hepatitis C Virus. Hepatol. Baltim. Md 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hu, L.; Dong, G.; Zhang, Y.; Ferreira da Silva-Júnior, E.; Liu, X.; Menéndez-Arias, L.; Zhan, P. Emerging Drug Design Strategies in Anti-Influenza Drug Discovery. Acta Pharm. Sin. B 2023, 13, 4715–4732. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Bannazadeh Baghi, H.; Sadri Nahand, J.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A New Approach to Covid-19 Therapy? Biomed. Pharmacother. Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F.; McConnell, S.; Casadevall, A. Very Low Levels of Remdesivir Resistance in SARS-COV-2 Genomes after 18 Months of Massive Usage during the COVID19 Pandemic: A GISAID Exploratory Analysis. Antiviral Res. 2022, 198, 105247. [Google Scholar] [CrossRef]
- Jahankhani, K.; Ahangari, F.; Adcock, I.M.; Mortaz, E. Possible Cancer-Causing Capacity of COVID-19: Is SARS-CoV-2 an Oncogenic Agent? Biochimie 2023, 213, 130–138. [Google Scholar] [CrossRef]
- Fugl, A.; Andersen, C.L. Epstein-Barr Virus and Its Association with Disease—A Review of Relevance to General Practice. BMC Fam. Pract. 2019, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Hartog, N.L.; Chesla, D.; Faber, W.; Love, C.P.; Karam, R.; Abualkheir, N.; Feldmann, B.; Teng, L.; McBride, T.; et al. High-Density Blood Transcriptomics Reveals Precision Immune Signatures of SARS-CoV-2 Infection in Hospitalized Individuals. Front. Immunol. 2021, 12, 694243. [Google Scholar] [CrossRef] [PubMed]
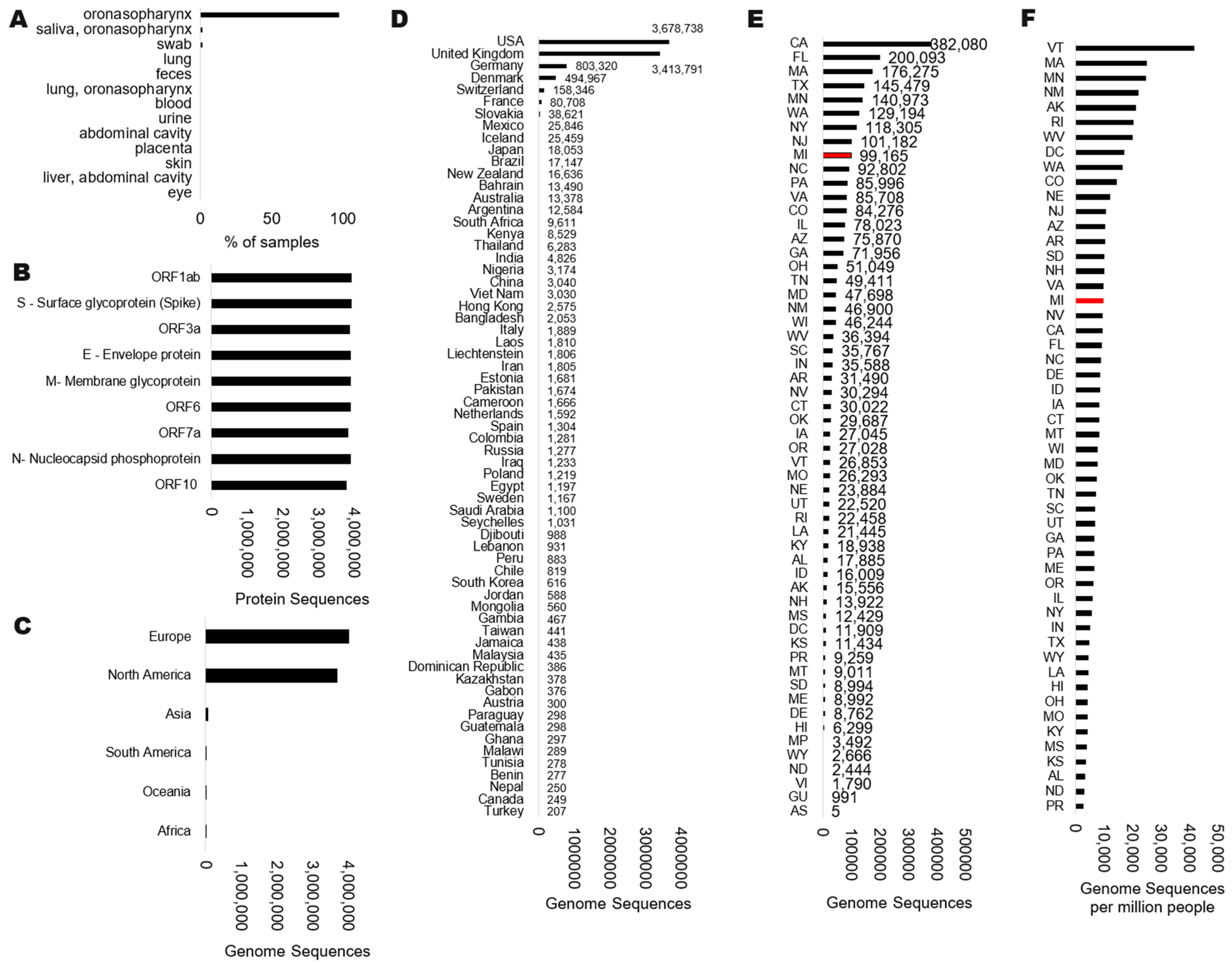


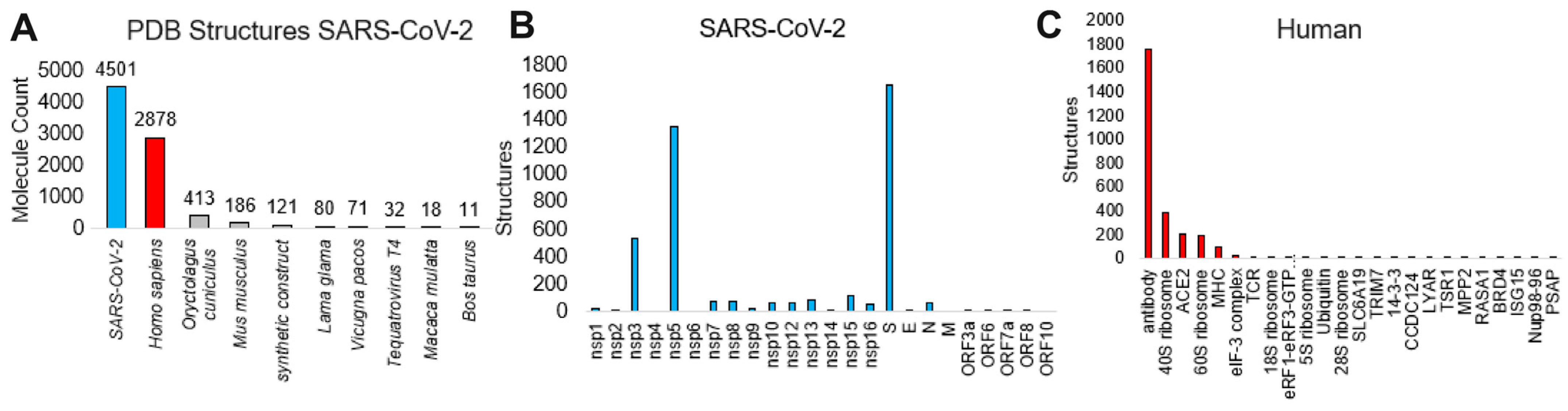
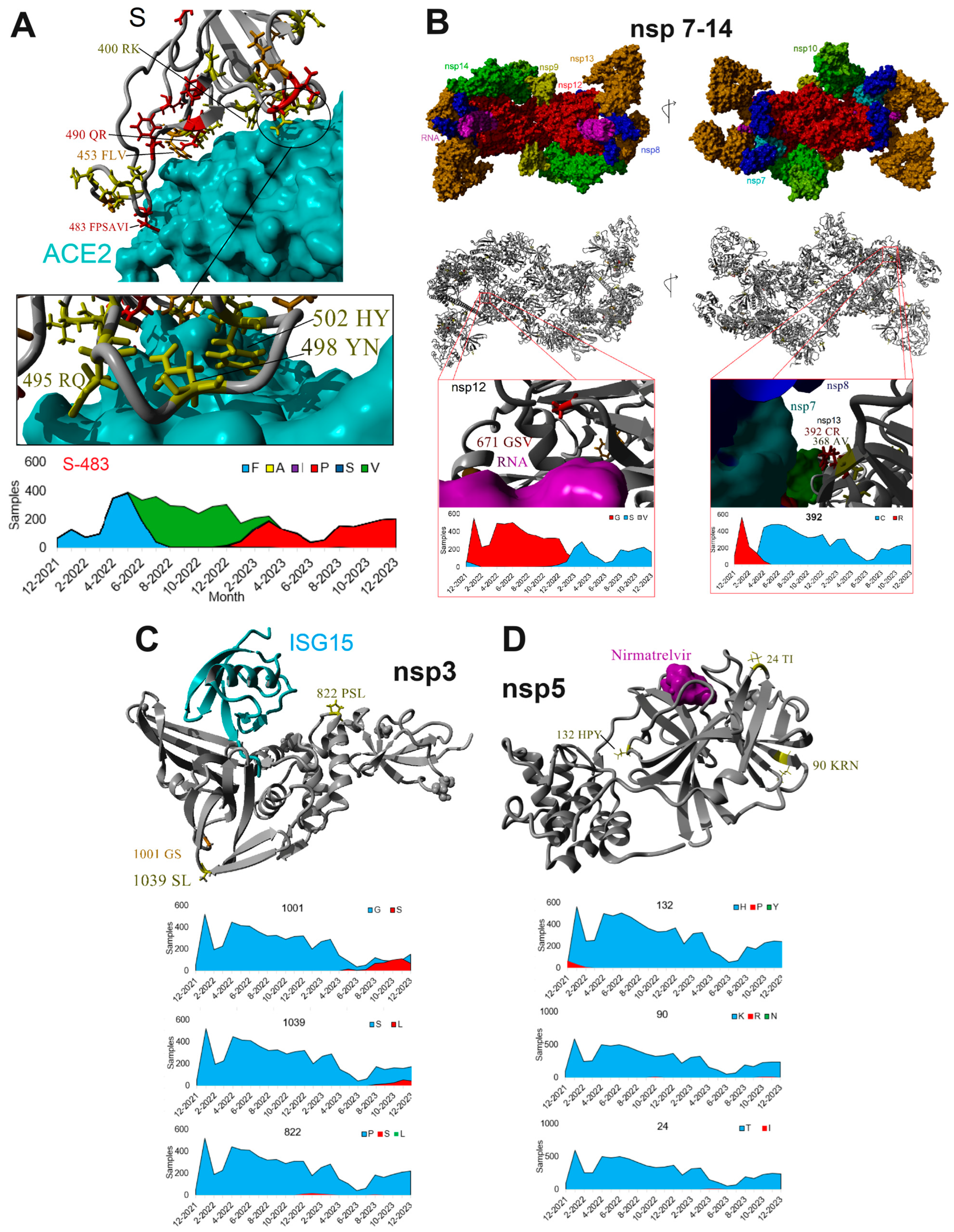

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Complete Sequences for Protein | Amino Acid Number | Consensus | Amino Acids Used | Amino Acids Used | % Drift |
|---|---|---|---|---|---|---|
| surface glycoprotein | 5102 | 483 | F | 6 | FPSAVI | 73.11 |
| membrane glycoprotein | 3617 | 3 | D | 4 | DHNG | 62.37 |
| surface glycoprotein | 5102 | 449 | R | 5 | RLMWQ | 53.21 |
| ORF6 | 7405 | 61 | L | 5 | LIDFP | 49.99 |
| surface glycoprotein | 5102 | 66 | H | 5 | H-YFS | 47.53 |
| surface glycoprotein | 5102 | 67 | V | 3 | V-I | 47.43 |
| surface glycoprotein | 5102 | 343 | R | 5 | RTISK | 46.37 |
| NSP4 | 7365 | 438 | L | 3 | LFY | 46.06 |
| surface glycoprotein | 5102 | 457 | N | 5 | NKISY | 41.40 |
| surface glycoprotein | 5102 | 141 | Y | 2 | Y- | 36.50 |
| surface glycoprotein | 5102 | 443 | G | 5 | GSTDV | 35.59 |
| surface glycoprotein | 5102 | 210 | G | 4 | GERV | 34.77 |
| surface glycoprotein | 5102 | 336 | D | 5 | DHVYG | 33.83 |
| surface glycoprotein | 5102 | 442 | V | 8 | VPSHALIF | 31.145 |
| surface glycoprotein | 5102 | 143 | H | 7 | HQK-YPL | 29.60 |
| surface glycoprotein | 5102 | 487 | F | 3 | FSP | 29.38 |
| surface glycoprotein | 5102 | 80 | V | 2 | VA | 29.09 |
| surface glycoprotein | 5102 | 365 | L | 2 | LI | 29.09 |
| surface glycoprotein | 5102 | 180 | Q | 6 | QEVHKL | 28.68 |
| NSP12 | 7233 | 671 | G | 3 | GSV | 28.61 |
| surface glycoprotein | 5102 | 249 | G | 3 | GVD | 26.81 |
| envelope protein | 7437 | 11 | T | 3 | TAM | 26.45 |
| NSP1 | 6468 | 47 | K | 2 | KR | 25.57 |
| surface glycoprotein | 5102 | 490 | Q | 2 | QR | 24.52 |
| NSP13 | 7231 | 36 | S | 2 | SP | 24.31 |
| NSP1 | 6468 | 135 | R | 4 | RSKN | 18.69 |
| NSP3 | 6434 | 1892 | A | 3 | ATG | 17.28 |
| NSP4 | 7365 | 264 | F | 2 | FL | 17.05 |
| NSP4 | 7365 | 327 | I | 4 | ITVF | 17.03 |
| ORF3a | 7465 | 223 | I | 2 | IT | 17.03 |
| nucleocapsid phosphoprotein | 7016 | 410 | R | 5 | RLSHC | 17.00 |
| NSP15 | 7426 | 112 | I | 3 | ITN | 16.89 |
| NSP13 | 7231 | 392 | C | 2 | CR | 16.60 |
| NSP3 | 6434 | 24 | I | 2 | IT | 16.52 |
| NSP3 | 6434 | 489 | S | 2 | SG | 16.47 |
| NSP3 | 6434 | 38 | K | 2 | KR | 15.99 |
| NSP3 | 6434 | 1265 | S | 2 | S- | 15.96 |
| NSP3 | 6434 | 1266 | L | 3 | LIV | 15.96 |
| NSP6 | 7472 | 105 | L | 2 | LF | 15.91 |
| NSP6 | 7472 | 186 | I | 2 | IV | 15.79 |
| NSP6 | 7472 | 257 | L | 3 | LFH | 12.17 |
| surface glycoprotein | 5102 | 441 | K | 5 | KTNRM | 11.00 |
| surface glycoprotein | 5102 | 453 | F | 3 | FLV | 10.80 |
| PubMed (All) | PubMed (Since 2020) | Google Scholar (All) | Google Scholar (Since 2020) | Google (All) | Ratio Google to PubMed (Since 2020) | |
|---|---|---|---|---|---|---|
| surface glycoprotein | 788,636 | 92,484 | 1,840,000 | 26,700 | 45,000,000 | 487 |
| Membrane glycoprotein | 786,289 | 91,863 | 1,920,000 | 24,200 | 38,400,000 | 418 |
| COVID-19 | 438,172 | 292,493 | 5,040,000 | 1,020,000 | 6,400,000,000 | 21,881 |
| pandemic | 274,892 | 171,320 | 4,220,000 | 683,000 | 1,510,000,000 | 8814 |
| SARS-CoV-2 | 231,386 | 186,717 | 2,500,000 | 781,000 | 459,000,000 | 2458 |
| viral genome | 117,243 | 19,241 | 4,220,000 | 30,400 | 120,000,000 | 6237 |
| viral genotype | 64,224 | 10,674 | 2,130,000 | 16,700 | 23,500,000 | 2202 |
| Envelope protein | 52,391 | 5939 | 2,110,000 | 23,900 | 46,300,000 | 7796 |
| RNA-directed RNA polymerase | 38,009 | 4142 | 35,100 | 7190 | 40,200,000 | 9705 |
| Helicase | 36,622 | 7834 | 332,000 | 25,200 | 19,500,000 | 2489 |
| Nucleocapsid | 28,311 | 5146 | 208,000 | 27,500 | 7,970,000 | 1549 |
| surface glycoprotein spike | 9146 | 2269 | 91,900 | 18,200 | 1,210,000 | 533 |
| papain like protease | 2031 | 574 | 95,100 | 17,000 | 776,000 | 1352 |
| 3C like protease | 1580 | 986 | 850,000 | 16,300 | 8,810,000 | 8935 |
| Nucleocapsid phosphoprotein | 1452 | 694 | 19,700 | 6350 | 131,000 | 189 |
| nsp2 | 1180 | 302 | 20,800 | 8450 | 459,000 | 1520 |
| nsp1 | 1124 | 324 | 26,600 | 12,400 | 428,000 | 1321 |
| nsp3 | 1049 | 377 | 20,500 | 11,700 | 269,000 | 714 |
| nsp4 | 949 | 203 | 17,000 | 6880 | 208,000 | 1025 |
| ORF1ab | 676 | 448 | 17,900 | 17,400 | 307,000 | 685 |
| ORF6 | 542 | 131 | 16,700 | 7070 | 192,000 | 1466 |
| nsp12 | 483 | 329 | 10,600 | 8830 | 470,000 | 1429 |
| nsp5 | 478 | 174 | 11,600 | 6780 | 212,000 | 1218 |
| ORF3a | 380 | 240 | 8840 | 8040 | 145,000 | 604 |
| nsp14 | 301 | 169 | 7940 | 6430 | 235,000 | 1391 |
| nsp13 | 256 | 154 | 7280 | 6080 | 540,000 | 3506 |
| nsp10 | 254 | 136 | 6640 | 4640 | 176,000 | 1294 |
| 2′-O-methyltransferase | 237 | 65 | 130,000 | 18,500 | 16,800,000 | 258,462 |
| nsp16 | 230 | 123 | 6880 | 5690 | 169,000 | 1374 |
| nsp15 | 215 | 118 | 6030 | 4910 | 224,000 | 1898 |
| nsp7 | 205 | 114 | 6920 | 5260 | 189,000 | 1658 |
| nsp9 | 200 | 96 | 5350 | 3500 | 189,000 | 1969 |
| nsp8 | 191 | 116 | 6850 | 5170 | 136,000 | 1172 |
| ORF10 | 190 | 67 | 8350 | 4450 | 73,900 | 1103 |
| ORF7a | 189 | 114 | 5350 | 4830 | 96,300 | 845 |
| Guanine-N7 methyltransferase | 175 | 34 | 2230 | 1070 | 53,700 | 1579 |
| nsp6 | 170 | 89 | 7660 | 4880 | 152,000 | 1708 |
| Uridylate-specific endoribonuclease | 86 | 58 | 745 | 480 | 17,200 | 297 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokop, J.W.; Alberta, S.; Witteveen-Lane, M.; Pell, S.; Farag, H.A.; Bhargava, D.; Vaughan, R.M.; Frisch, A.; Bauss, J.; Bhatti, H.; et al. SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins. Microorganisms 2024, 12, 1863. https://doi.org/10.3390/microorganisms12091863
Prokop JW, Alberta S, Witteveen-Lane M, Pell S, Farag HA, Bhargava D, Vaughan RM, Frisch A, Bauss J, Bhatti H, et al. SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins. Microorganisms. 2024; 12(9):1863. https://doi.org/10.3390/microorganisms12091863
Chicago/Turabian StyleProkop, Jeremy W., Sheryl Alberta, Martin Witteveen-Lane, Samantha Pell, Hosam A. Farag, Disha Bhargava, Robert M. Vaughan, Austin Frisch, Jacob Bauss, Humza Bhatti, and et al. 2024. "SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins" Microorganisms 12, no. 9: 1863. https://doi.org/10.3390/microorganisms12091863
APA StyleProkop, J. W., Alberta, S., Witteveen-Lane, M., Pell, S., Farag, H. A., Bhargava, D., Vaughan, R. M., Frisch, A., Bauss, J., Bhatti, H., Arora, S., Subrahmanya, C., Pearson, D., Goodyke, A., Westgate, M., Cook, T. W., Mitchell, J. T., Zieba, J., Sims, M. D., ... Caulfield, A. J. (2024). SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins. Microorganisms, 12(9), 1863. https://doi.org/10.3390/microorganisms12091863