
Are Changes Occurring in Bacterial Taxa Community and Diversity with the Utilization of Different Substrates within SIR Measurements?

Abstract
:1. Introduction
2. Materials and Method
2.1. Study Site and Soil Collection
2.2. Substrate Utilization
2.3. DNA Extraction, Amplicon Sequencing, and Sequence Processing
3. Data Analysis
Statistical Analyses
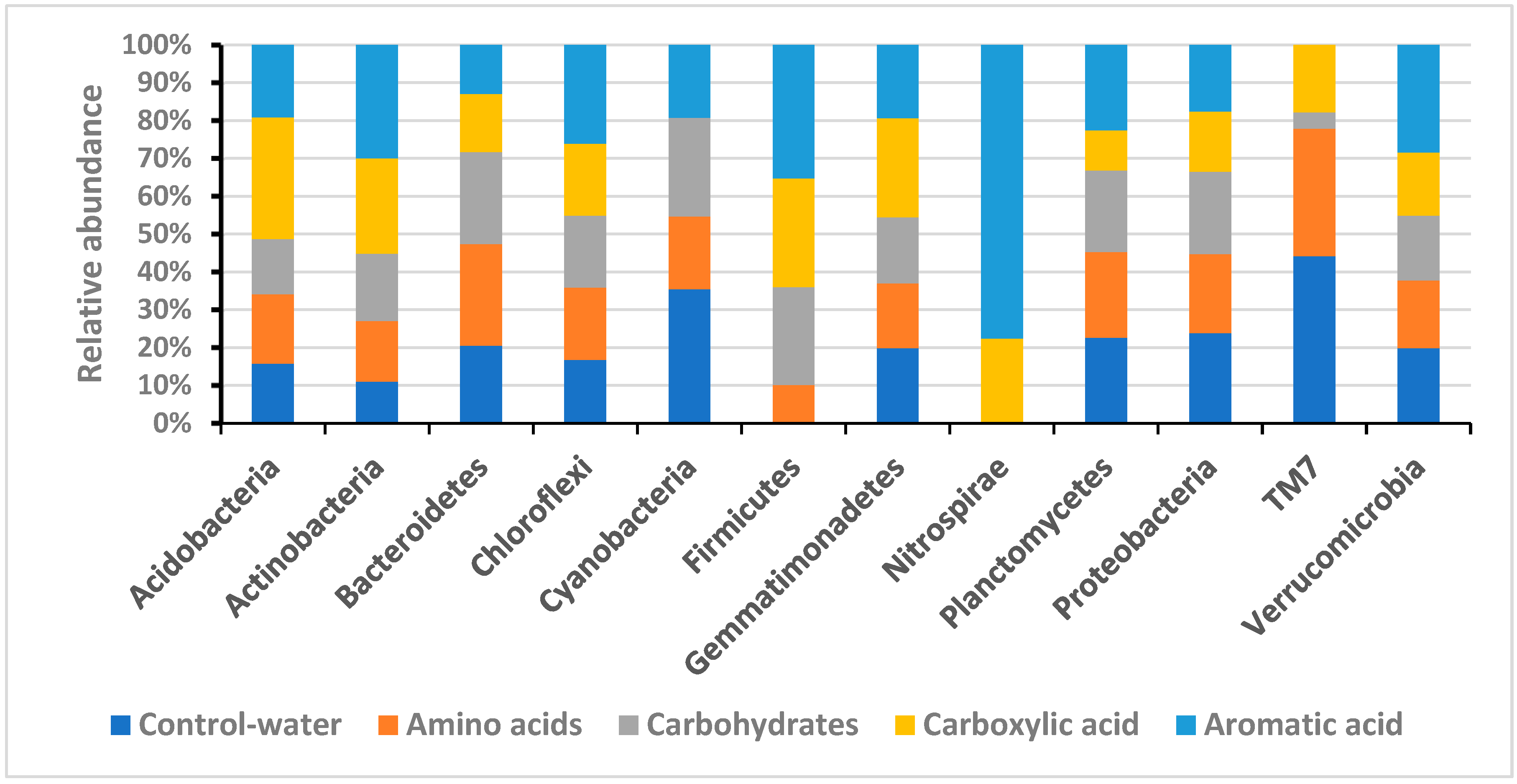
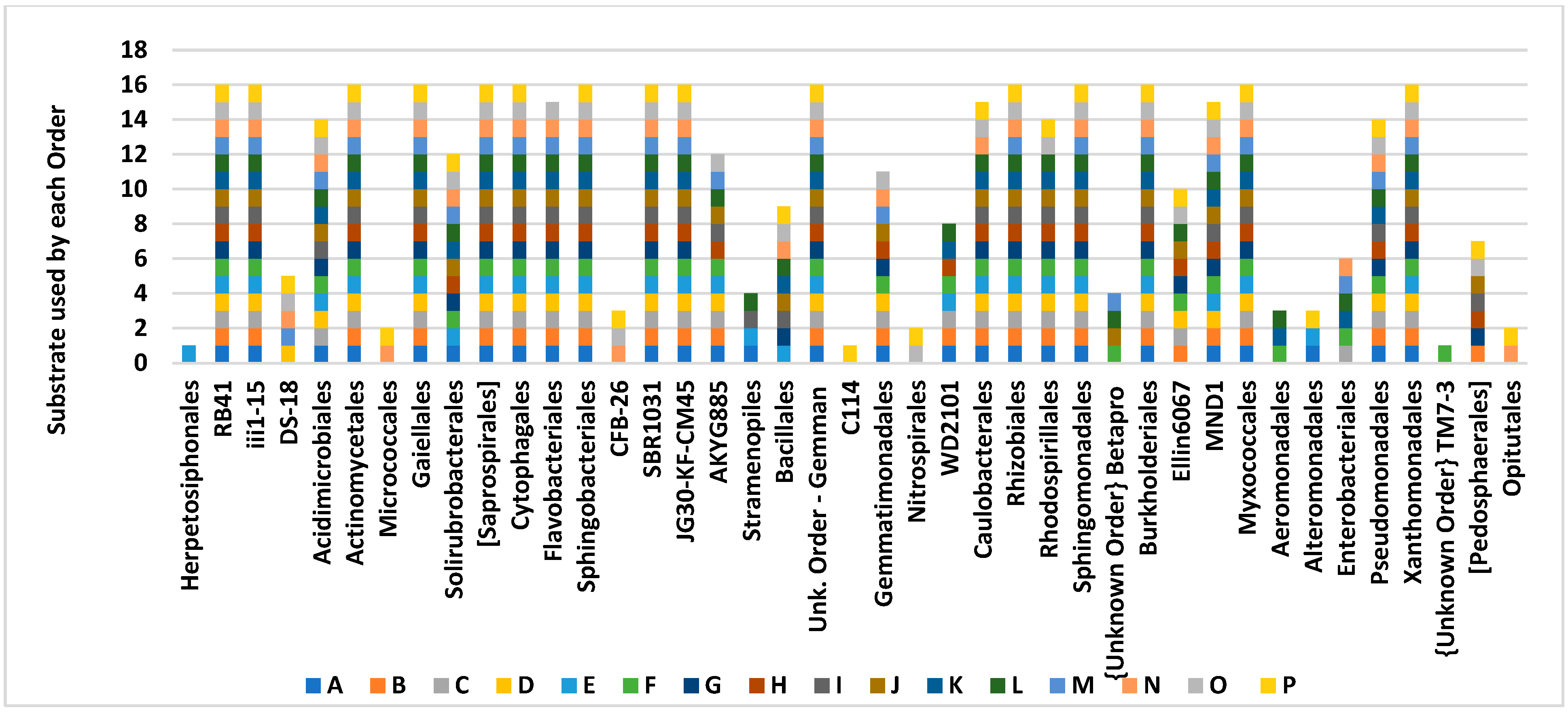
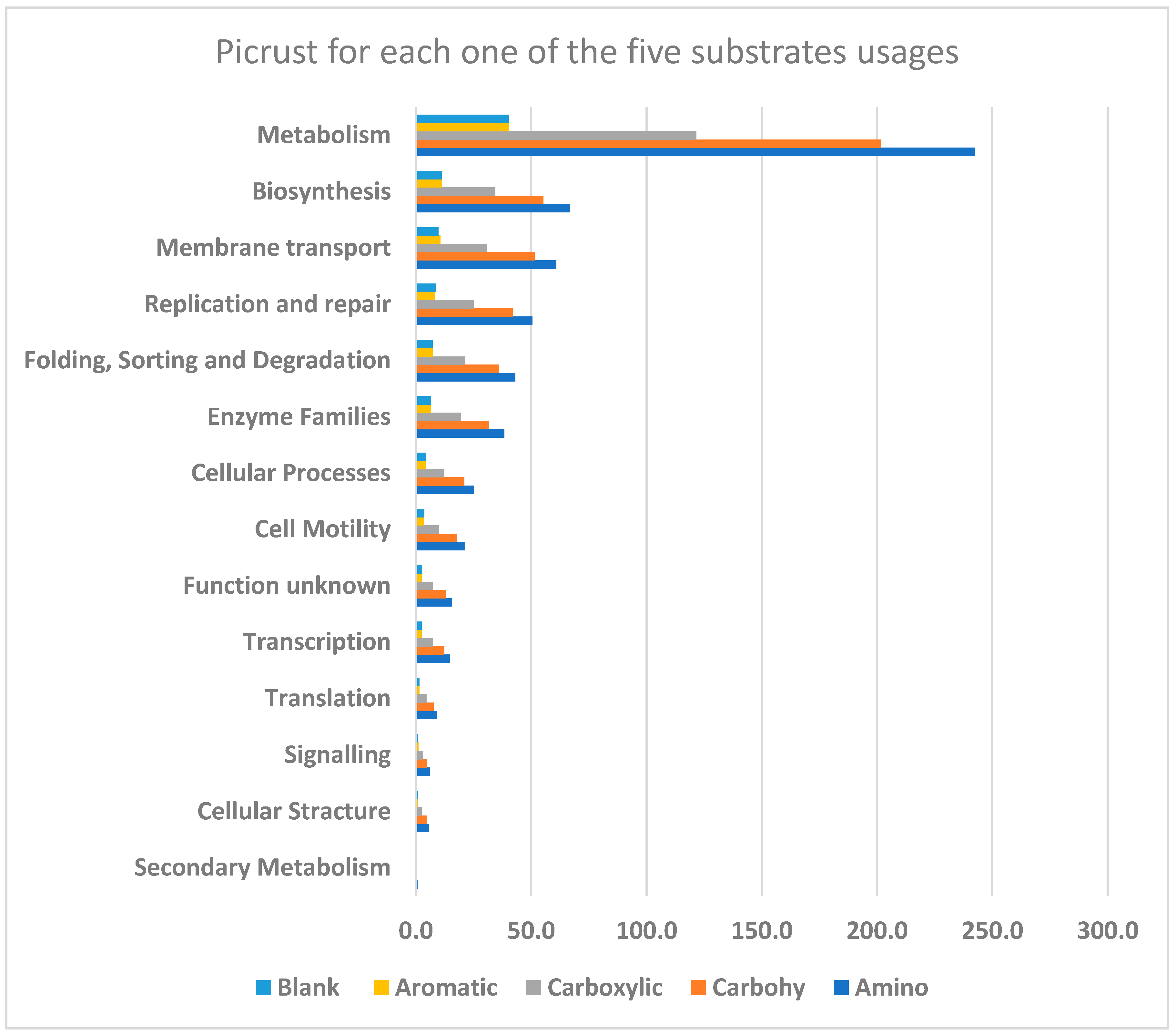
4. Results
Soil Bacteria
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Noy-Meir, I. Desert ecosystems: Environment and producers. Annu. Rev. Ecol. Syst. 1973, 4, 25–51. [Google Scholar] [CrossRef]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.J.; Heal, O.W.; Anderson, J.M. Decomposition in Terrestrial Ecosystems; Blackwell: Oxford, UK, 1979. [Google Scholar]
- Anderson, J.M.; Rayner, A.D.M.; Walton, D.W.H. Invertebrate-Microbial Interactions; Cambridge University Press: Cambridge, UK, 1984. [Google Scholar]
- Maraun, M.; Scheu, S. Seasonal changes in microbial biomass and activity in leaf litter layers of beech (Fagus sylvatica) forests on a basalt-limestone gradient. Pedobiologia 1996, 40, 21–31. [Google Scholar] [CrossRef]
- Anderson, J.P.E.; Domsch, K.H. Quantities of plant nutrients in the microbial biomass of selected soil. Soil. Sci. 1980, 130, 211–216. [Google Scholar] [CrossRef]
- Christensen, H.; Funck-Jensen, D. Growth Rate of Rhizosphere Bacteria Measured Directly by the Tritiated Thymldine Incorporation Technique. Soil. Biol. Biochem. 1989, 21, 113–117. [Google Scholar] [CrossRef]
- Bünemann, E.K.; Bossio, D.A.; Smithson, P.C.; Frossard, E.; Oberson, A. Microbial community composition and substrate use in a highly weathered soil as affected by crop rotation and P fertilization. Soil Biol. Biochem. 2004, 36, 889–901. [Google Scholar] [CrossRef]
- Rousk, J.; Baath, E. Fungal and bacterial growth in soil with plant materials of different C/N ratios. FEMS Microbiol. Ecol. 2007, 62, 258–267. [Google Scholar] [CrossRef]
- Bossio, D.A.; Scow, K.M.; Gunapala, N.; Graham, K.J. Determinants of soil microbial communities: Effects of agricultural management, season, and soil type on phospholipid fatty acid profiles. Microb. Ecol. 1998, 36, 1–12. [Google Scholar] [CrossRef]
- Steinberger, Y.; Zelles, L.; Bai, Q.Y.; von Lutzow, M.; Munch, J.C. Phospholipid fatty acid profiles as indicators for the microbial community structure in soils along a climatic transect in the Judean Desert. Biol. Fertil. Soils 1999, 28, 292–300. [Google Scholar] [CrossRef]
- Fliessbach, A.; Ma¨der, P.; Niggli, U. Mineralization and microbial assimilation of 14C-labeled straw in soils of organic and conventional agricultural systems. Soil. Biol. Biochem. 2000, 32, 1131–1139. [Google Scholar] [CrossRef]
- Vishnevetsky, S.; Steinberger, Y. Bacterial and fungal dynamics and their contribution to microbial biomass in desert soil. J. Arid. Environ. 1997, 37, 83–90. [Google Scholar] [CrossRef]
- Oren, A.; Steinberger, Y. Catabolic profiles of soil fungal communities along a geographic climatic gradient in Israel. Soil. Biol. Biochem. 2008, 40, 2578–2587. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.N.; Rushton, S.P.; Lanyon, C.V.; Whiteley, A.S.; Waite, I.S.; Brookes, P.C.; Kemmitt, S.; Evershed, R.P.; O’Donnell, A.G. Taxon-specific responses of soil bacteria to the addition of low level C inputs. Soil Biol. Biochem. 2010, 42, 1624–1631. [Google Scholar] [CrossRef]
- Ai, C.; Liang, G.; Sun, J.; Wang, X.; He, P.; Zhou, W.; He, X. Reduced dependence of rhizosphere microbiome on plant-derived carbon in 32-year long-term inorganic and organic fertilized soils. Soil Biol. Biochem. 2015, 80, 70–78. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Liu, Y.; Jia, Z.; Li, H.; Yang, Y.; Wang, D.; He, H.; Zhang, X. Identification of microbial strategies for labile substrate utilization at phylogenetic classification using a microcosm approach. Soil Biol. Biochem. 2021, 153, 107970. [Google Scholar] [CrossRef]
- Brock, T.D. Effect of water potential on a microcoleus (Cyanophyceae) from a desert crust. J. Phycol. 1975, 11, 316–320. [Google Scholar] [CrossRef]
- Sherman, C.; Unc, A.; Doniger, T.; Ehrlich, R.; Steinberger, Y. The effect of human trampling activity on a soil microbial community at the Oulanka Natural Reserve, Finland. Appl. Soil. Ecol. 2018, 135, 104–112. [Google Scholar] [CrossRef]
- Levi, M.; Applebaum, I.; Sherman, C.; Doniger, T.; Steinberger, Y. Soil fungal community of wheat Triticum aestivum rhizosphere at different phenological stages under a rain-fed management. Rhizosphere 2022, 24, 100605. [Google Scholar] [CrossRef]
- Oren, A.; Steinberger, Y. Coping with artifacts induced by CaCO3-CO2-H2O equilibria in substrate utilization profiling of calcareous soils. Soil. Biol. Biochem. 2008, 40, 2569–2577. [Google Scholar] [CrossRef]
- McDonald, M.J.; Wang, W.C.; Huang, H.D.; Leu, J.Y. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011, 9, e1000622. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.; Reynolds, J.F. Statistical Ecology: A Primer in Methods and Computing; John Wiley & Sons: Hoboken, NJ, USA, 1988; Volume 1. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Morlat, R.; Chaussod, R. Long-term Additions of Organic Amendments in a Loire Valley Vineyard. I. Effects on Properties of a Calcareous Sandy Soil. Am. J. Enol. Vitic. 2008, 59, 353–363. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Dumont, M.G.; Murrell, J.C. Stable isotope probing—Linking microbial identity to function. Nat. Rev. Microbiol. 2005, 3, 499–504. [Google Scholar] [CrossRef]
- Neufeld, J.D.; Dumont, M.G.; Vohra, J.; Murrell, J.C. Methodological considerations for the use of stable isotope probing in microbial ecology. Microb. Ecol. 2007, 53, 435–442. [Google Scholar] [CrossRef]
- Strickland, M.S.; Wickings, K.; Bradford, M.A. The fate of glucose, a low molecular weight compound of root exudates, in the belowground food web of forests and pastures. Soil Biol. Biochem. 2012, 49, 23–29. [Google Scholar] [CrossRef]
- Koch, A.L. Oligotrophs versus copiotrophs. Bioessays 2001, 23, 657–661. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Padmanabhan, S.; DeRito, C.; Gray, A.; Gannon, D.; Snape, J.R.; Tsai, C.S.; Park, W.; Jeon, C.; Madsen, E.L. Respiration of 13C-labeled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13C-labeled soil DNA. Appl. Environ. Microbiol. 2003, 69, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Verastegui, Y.; Cheng, J.; Engel, K.; Kolczynski, D.; Mortimer, S.; Lavigne, J.; Montalibet, J.; Romantsov, T.; Hall, M.; McConkey, B.J.; et al. Multisubstrate isotope labeling and metagenomic analysis of active soil bacterial communities. MBio 2014, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.; Dibbern, D.; Moll, J.; Huenninghaus, M.; Koller, R.; Krueger, D.; Marhan, S.; Urich, T.; Wubet, T.; Bonkowski, M.; et al. Resource partitioning between bacteria, fungi, and protists in the detritusphere of an agricultural soil. Front. Microbiol. 2016, 7, 1524. [Google Scholar] [CrossRef] [PubMed]
- Arcand, M.M.; Levy-Booth, D.J.; Helgason, B.L. Resource legacies of organic and conventional management differentiate soil microbial carbon use. Front. Microbiol. 2017, 8, 2293. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, C.; Ruan, Y.; Luo, G.; Wang, M.; Ling, N.; Shen, Q.; Guo, S. Are the microbial communities involved in glucose assimilation in paddy soils treated with different fertilization regimes for three years similar? J. Soils Sediments 2018, 18, 2476–2490. [Google Scholar] [CrossRef]
- Morrissey, E.M.; Mau, R.L.; Schwartz, E.; Caporaso, J.G.; Dijkstra, P.; Van Gestel, N.; Koch, B.J.; Liu, C.M.; Hayer, M.; McHugh, T.A.; et al. Phylogenetic organization of bacterial activity. ISME J. 2016, 10, 2336–2340. [Google Scholar] [CrossRef]
- Pepe-Ranney, C.; Campbell, A.N.; Koechli, C.N.; Berthrong, S.; Buckley, D.H. Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front. Microbiol. 2016, 7, 703. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Brabcová, V.; Štursová, M.; Davidová, A.; Jansa, J.; Cajthaml, T.; Baldrian, P. Decomposer food web in a deciduous forest shows high share of generalist microorganisms and importance of microbial biomass recycling. ISME J. 2018, 12, 1768–1778. [Google Scholar] [CrossRef]
- Campbell, C.D.; Chapman, S.J.; Cameron, C.M.; Davidson, M.S.; Potts, J.M. A rapid microtiter plate method to measure carbon dioxide evolved from carbon substrate amendments so as to determine the physiological profiles of soil microbial communities by using whole soil. Appl. Environ. Microbiol. 2003, 69, 3593–3599. [Google Scholar] [CrossRef]
- Carrara, F.; Giometto, A.; Seymour, M.; Rinaldo, A.; Altermatt, F. Inferring species interactions in ecological communities: A comparison of methods at different levels of complexity. Methods Ecol. Evol. 2015, 6, 895–906. [Google Scholar] [CrossRef]
- Cui, J.; Yang, B.; Zhang, M.; Song, D.; Xu, X.; Ai, C.; Liang, G.; Zhou, W. Investigating the effects of organic amendments on soil microbial composition and its linkage to soil organic carbon: A global meta-analysis. Sci. Total Environ. 2023, 894, 164899. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carboxylic Acids | Carbohydrates | Amino Acids | Aromatic Acids |
|---|---|---|---|
| Citric acid | L-Arabinose | L-Alanine | 3,4 Dihidroxybenzoic acid |
| L-Malic acid | D-Fructose | Arginine | |
| Oxalic acid | D-Glucose | L-Cysteine HCI | |
| Trehalose | Amino Butiric acid | ||
| D-Galactose | L-Lysine | ||
| N-acetyl-Glucosamine |
| id | p | E Value | FWER | q Value Factor | q Value |
|---|---|---|---|---|---|
| Proteobacteria | 1.36 × 10−5 | 0.00015 | 0.00015 | 11 | 0.00015 |
| Acidobacteria | 3.45 × 10−5 | 0.00038 | 0.000379 | 5.5 | 0.00019 |
| Bacteroidetes | 3.50 × 10−5 | 0.000385 | 0.000385 | 3.666667 | 0.000128 |
| Actinobacteria | 6.91 × 10−5 | 0.00076 | 0.00076 | 2.75 | 0.00019 |
| Planctomycetes | 3.46 × 10−3 | 0.038045 | 0.037394 | 2.2 | 0.007609 |
| Gemmatimonadetes | 3.59 × 10−3 | 0.039462 | 0.038762 | 1.833333 | 0.006577 |
| Cyanobacteria | 2.97 × 10−2 | 0.326927 | 0.28243 | 1.571429 | 0.046704 |
| Chloroflexi | 4.15 × 10−2 | 0.456824 | 0.372857 | 1.375 | 0.057103 |
| Nitrospirae | 6.44 × 10−2 | 0.708165 | 0.519049 | 1.222222 | 0.078685 |
| Firmicutes | 1.49 × 10−1 | 1.637842 | 0.830247 | 1.1 | 0.163784 |
| TM7 | 2.27 × 10−1 | 2.498659 | 0.941245 | 1 | 0.227151 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinberger, Y.; Doniger, T.; Applebaum, I.; Sherman, C. Are Changes Occurring in Bacterial Taxa Community and Diversity with the Utilization of Different Substrates within SIR Measurements? Microorganisms 2024, 12, 2034. https://doi.org/10.3390/microorganisms12102034
Steinberger Y, Doniger T, Applebaum I, Sherman C. Are Changes Occurring in Bacterial Taxa Community and Diversity with the Utilization of Different Substrates within SIR Measurements? Microorganisms. 2024; 12(10):2034. https://doi.org/10.3390/microorganisms12102034
Chicago/Turabian StyleSteinberger, Yosef, Tirza Doniger, Itaii Applebaum, and Chen Sherman. 2024. "Are Changes Occurring in Bacterial Taxa Community and Diversity with the Utilization of Different Substrates within SIR Measurements?" Microorganisms 12, no. 10: 2034. https://doi.org/10.3390/microorganisms12102034