
Gram Negative Biofilms: Structural and Functional Responses to Destruction by Antibiotic-Loaded Mixed Polymeric Micelles

 , , ,
, , ,  , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of PDMAEMA35-b-PCL70-b-PDMAEMA35 Triblock Copolymer
2.2. Preparation of MPMs
2.3. Loading of MPMs with Antibiotics
2.4. Calculation of Solubility and Polymer-Drug Miscibility Parameters
2.5. In Vitro Release of Antibiotics
2.6. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.7. Size Exclusion Chromatography (SEC)
2.8. High-Performance Liquid Chromatography (HPLC)
2.9. Dynamic and Electrophoretic Light Scattering (DLS and ELS)
2.10. Atomic Force Microscopy (AFM)
2.11. Cytotoxicity
2.12. Bacterial Strains and Cultivation
2.13. Biofilm Dispersion Experiments
2.14. Biofilm Cell Viability Estimation
2.15. Scanning Electron Microscopy (SEM)
2.16. Preparation of Murine Skin Explants
2.17. P. aeruginosa Biofilm Model on Ex Vivo Murine Skin Explants
2.18. Determination of NO Production by Ex Vivo Skin Explants
2.19. Secretion of IL-17A by Skin Explants
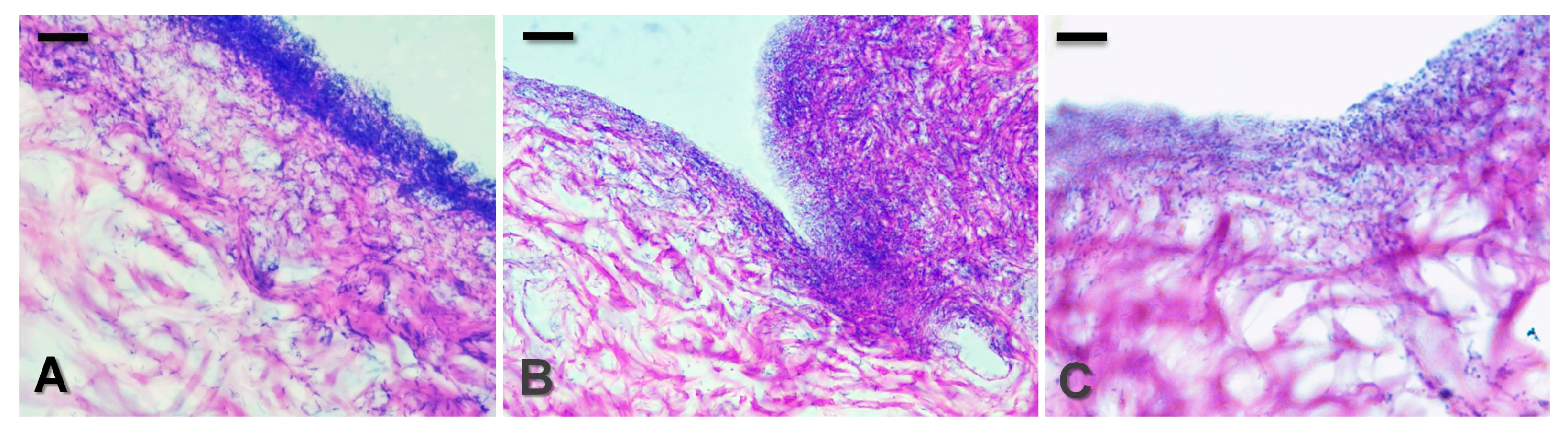
2.20. Histological Evaluation of Skin Explants
3. Results and Discussion
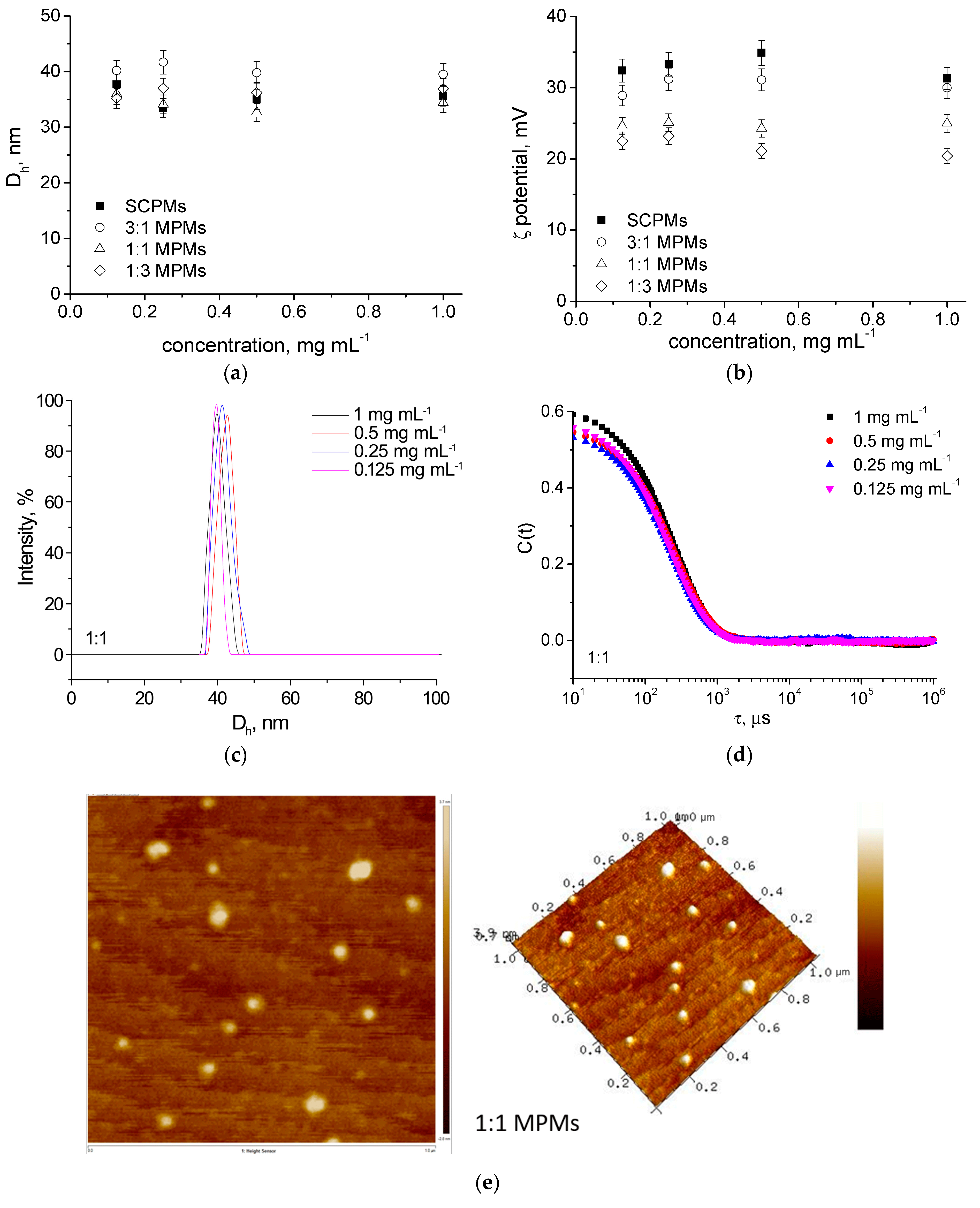
3.1. Preparation and Characterization of MPMs
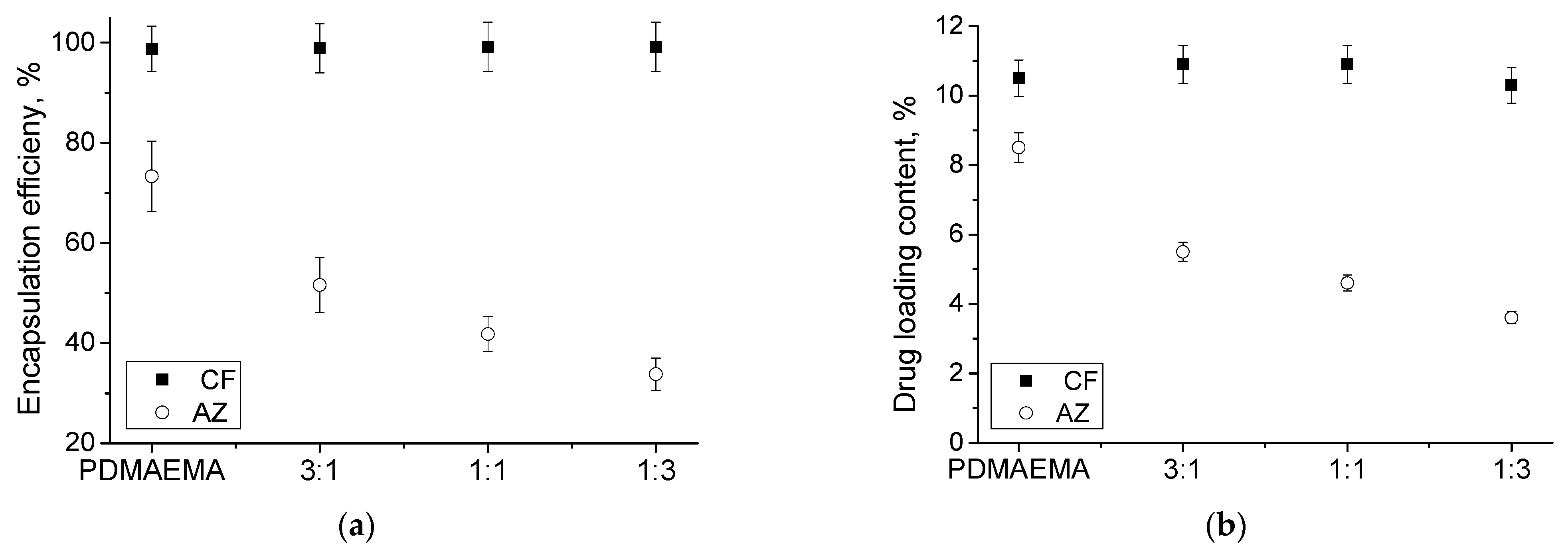
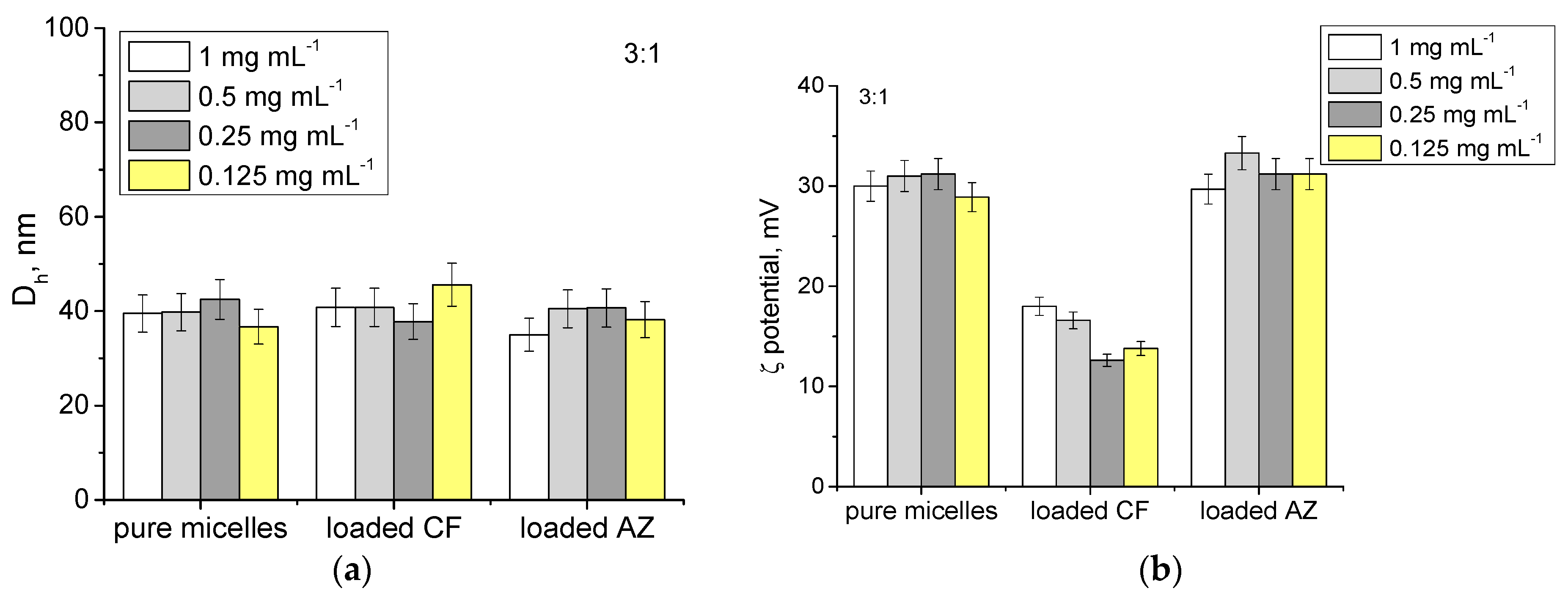
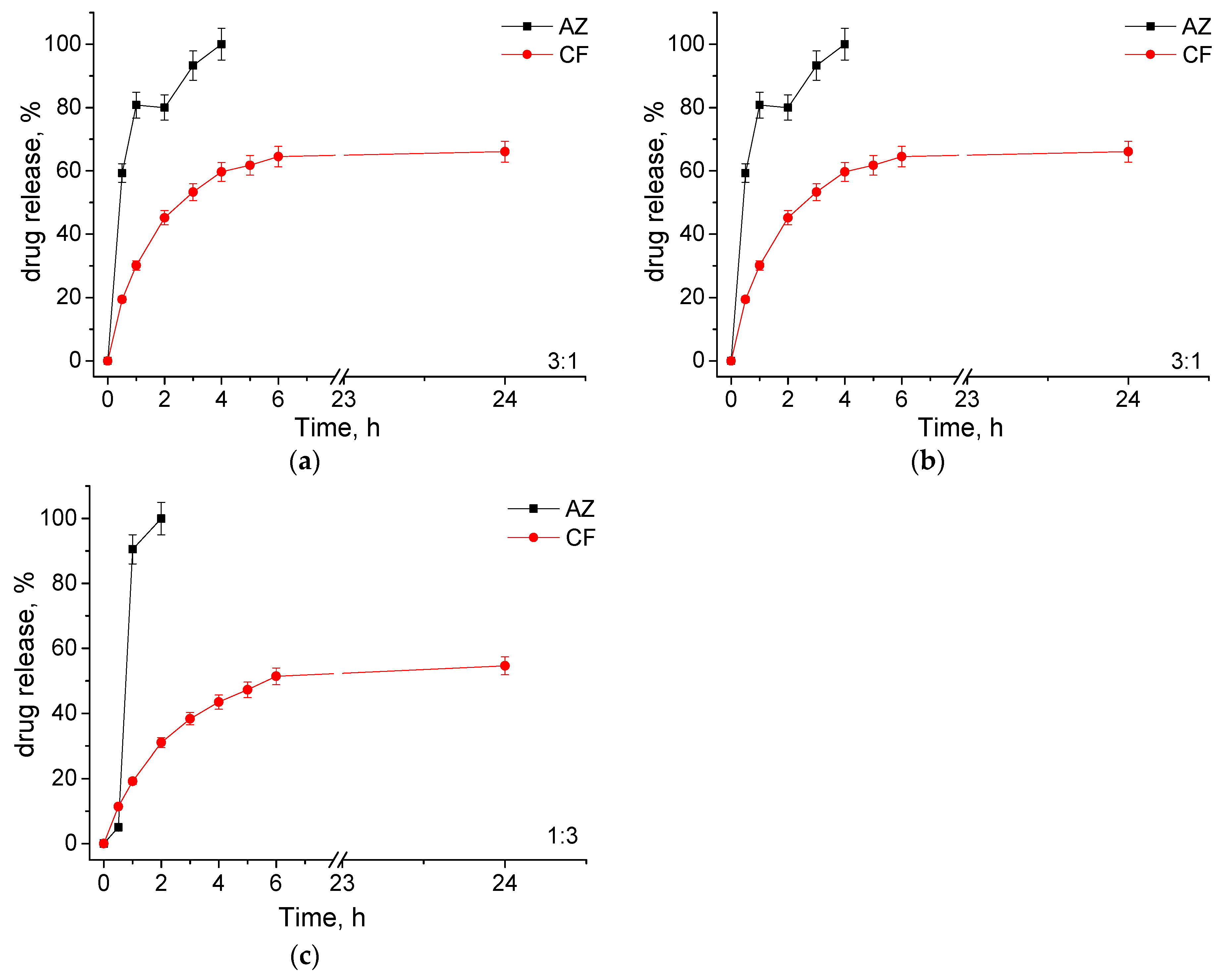
3.2. Loading of MPMs with Antibiotics and Release
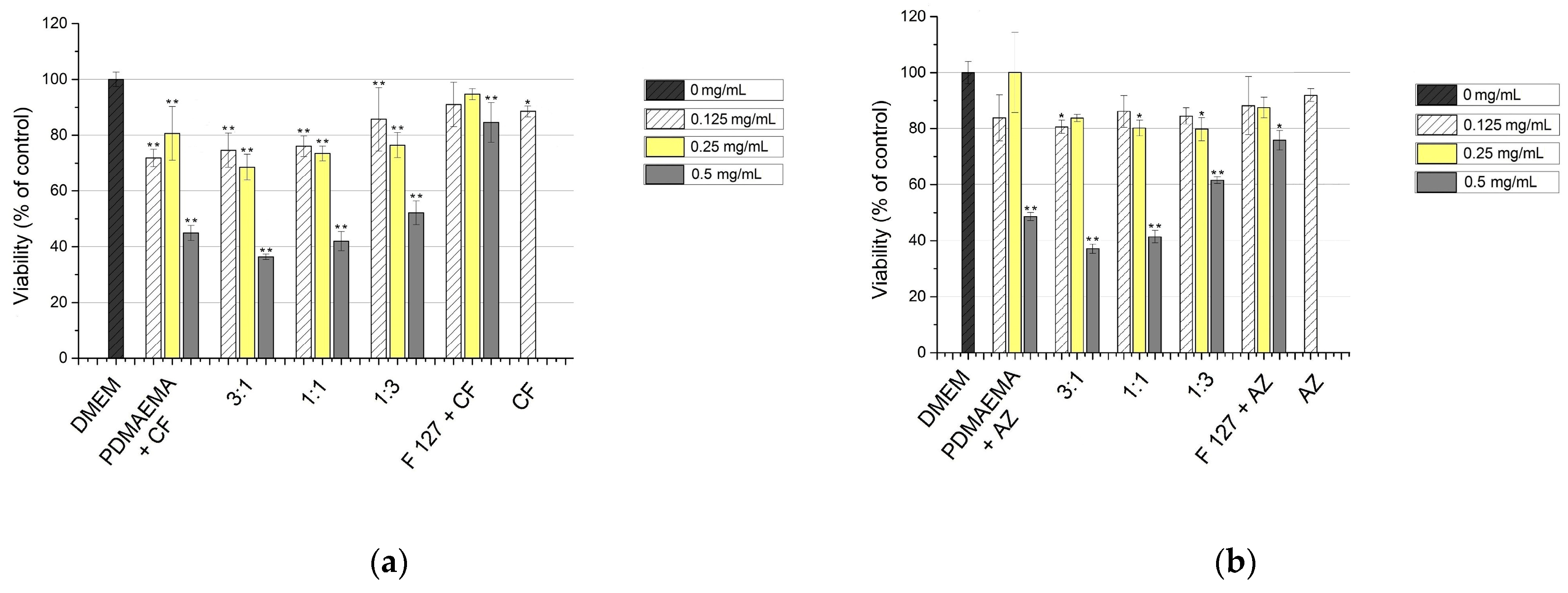
3.3. In Vitro Cytotoxicity
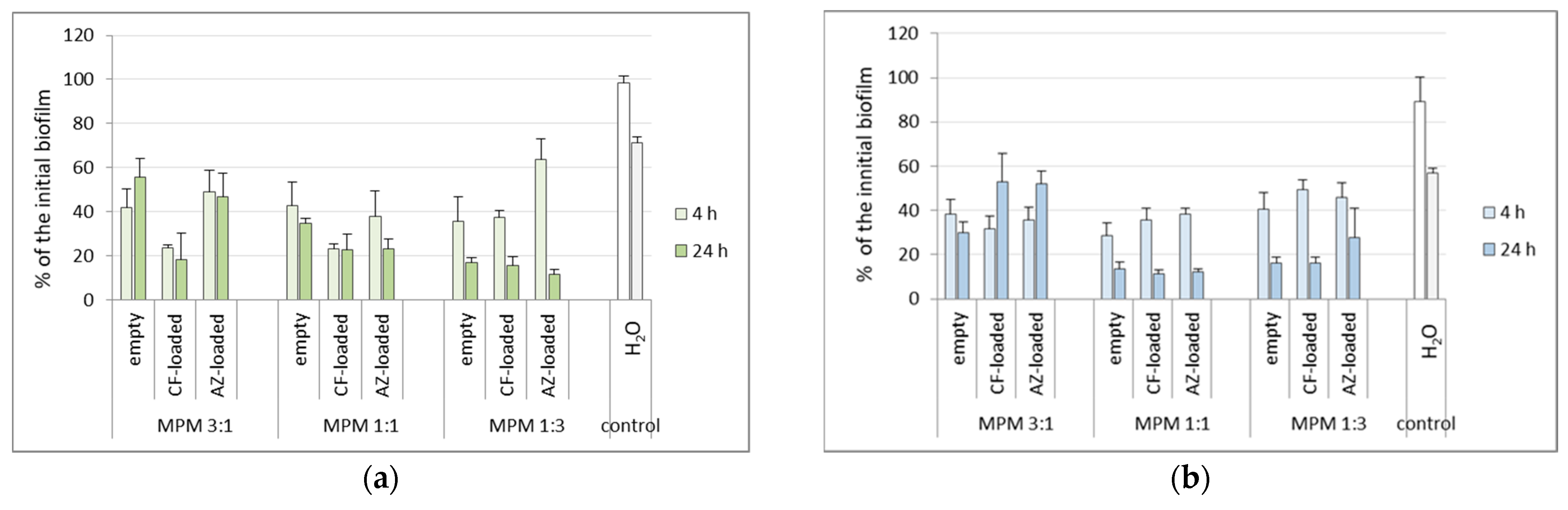
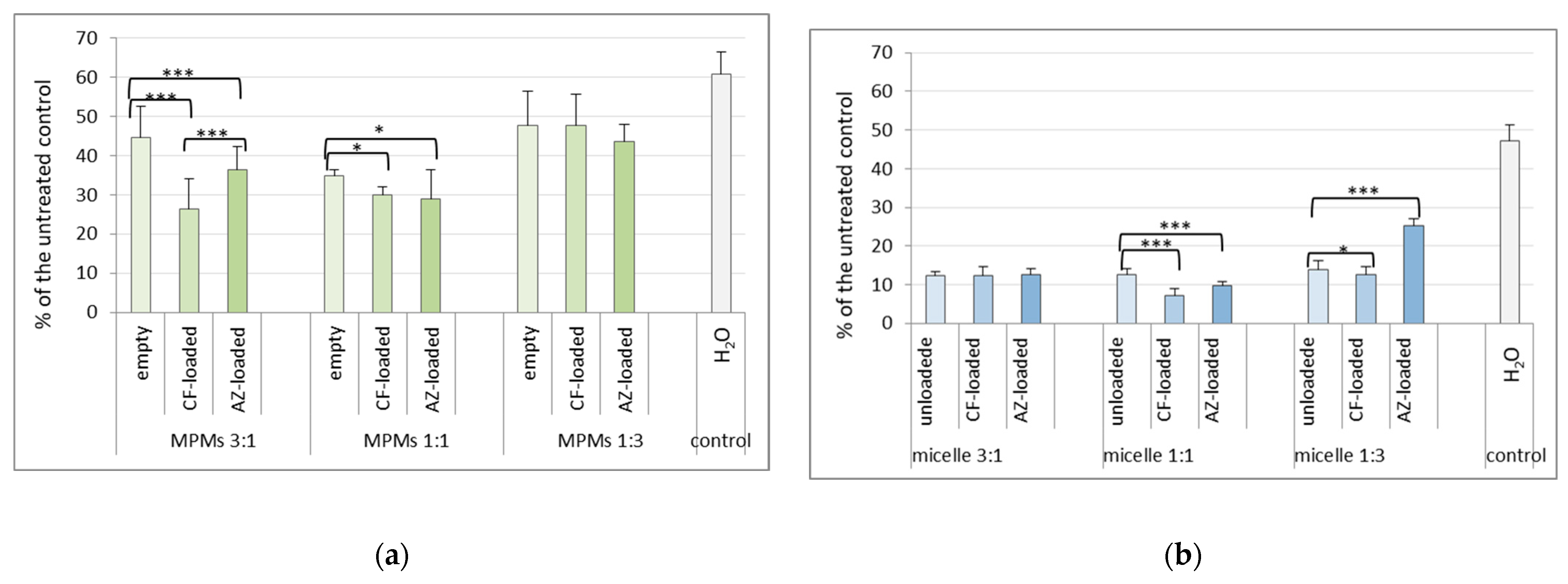
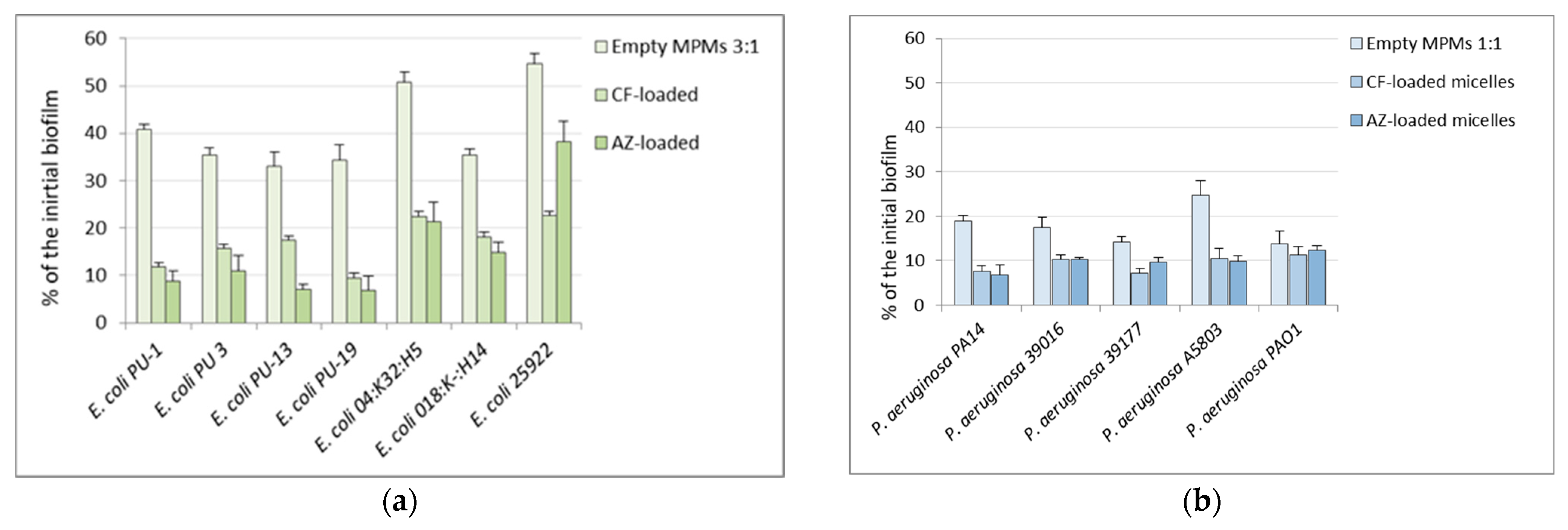
3.4. Effects of the Mixed Micelles on Pre-Formed 24 h Biofilms
3.5. Effects of MPMs on an Ex Vivo Skin Model of P. aeruginosa PAO1 Biofilm Infection
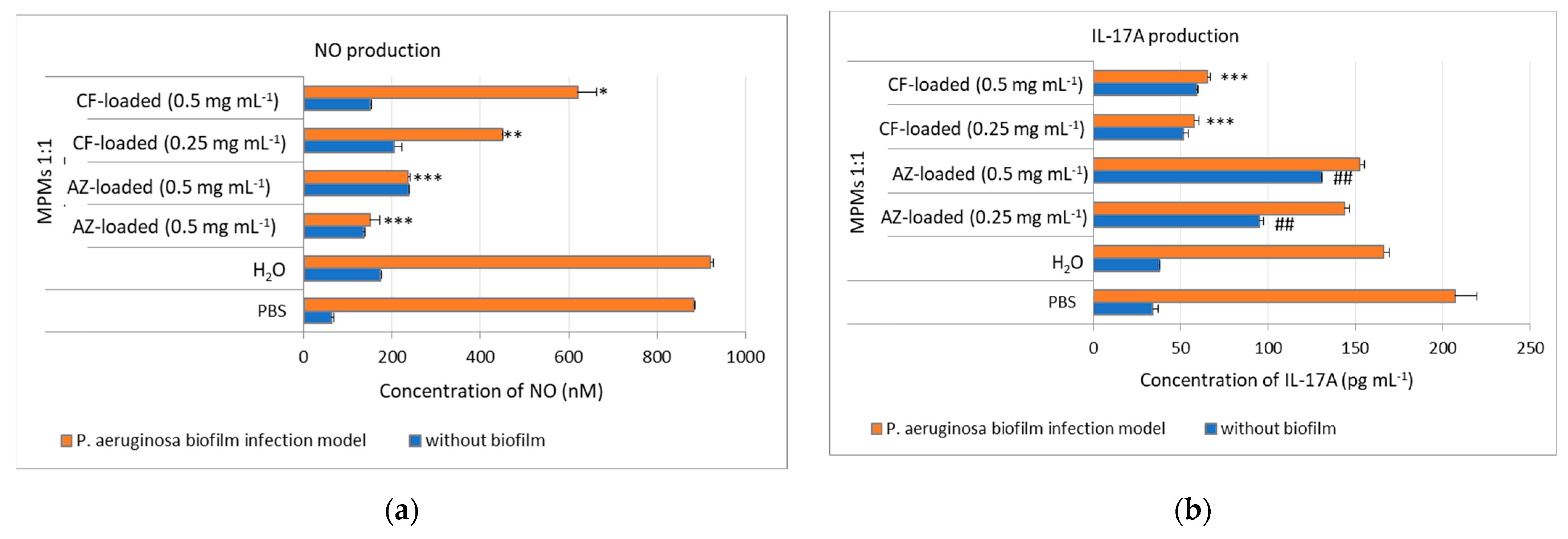
3.6. MPM 1:1 Loaded with CF Sustained NO Production in Ex Vivo Murine Skin Explant P. aeruginosa Biofilm Model
3.7. MPMs 1:1 Loaded with CF Inhibited IL-17A Production in Ex Vivo Murine Skin Explants P. aeruginosa Biofilm Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, R.M.; Soares, F.A.; Reis, S.; Nunes, C.; Van Dijck, P. Innovative Strategies Toward the Disassembly of the EPS Matrix in Bacterial Biofilms. Front. Microbiol. 2020, 11, 952. [Google Scholar] [CrossRef]
- Damyanova, T.; Dimitrova, P.D.; Borisova, D.; Topouzova-Hristova, T.; Haladjova, E.; Paunova-Krasteva, T. An Overview of Biofilm-Associated Infections and the Role of Phytochemicals and Nanomaterials in Their Control and Prevention. Pharmaceutics 2024, 16, 162. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, B.; Lin, N.; Gao, J. Recent Nanotechnology-Based Strategies for Interfering with the Life Cycle of Bacterial Biofilms. Biomater. Sci. 2023, 11, 1648–1664. [Google Scholar] [CrossRef] [PubMed]
- Ciofu, O.; Moser, C.; Jensen, P.Ø.; Høiby, N. Tolerance and Resistance of Microbial Biofilms. Nat. Rev. Microbiol. 2022, 20, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Stoitsova, S.; Paunova-Krasteva, T.; Dimitrova, P.D.; Damyanova, T. The Concept for the Antivirulence Therapeutics Approach as Alternative to Antibiotics: Hope or Still a Fiction? Biotechnol. Biotechnol. Equip. 2022, 36, 697–705. [Google Scholar] [CrossRef]
- Roshani, M.; Taheri, M.; Goodarzi, A.; Yosefimashouf, R.; Shokoohizadeh, L. Evaluation of Antibiotic Resistance, Toxin-Antitoxin Systems, Virulence Factors, Biofilm-Forming Strength and Genetic Linkage of Escherichia coli Strains Isolated from Bloodstream Infections of Leukemia Patients. BMC Microbiol. 2023, 23, 327. [Google Scholar] [CrossRef]
- Kolpen, M.; Kragh, K.N.; Enciso, J.B.; Faurholt-Jepsen, D.; Lindegaard, B.; Egelund, G.B.; Jensen, A.V.; Ravn, P.; Mathiesen, I.H.M.; Gheorghe, A.G.; et al. Bacterial Biofilms Predominate in Both Acute and Chronic Human Lung Infections. In Proceedings of the 10.01—Respiratory Infections and Bronchiectasis; European Respiratory Society: Lausanne, Switzerland, 2022; p. 4. [Google Scholar]
- Kolpen, M.; Jensen, P.Ø.; Faurholt-Jepsen, D.; Bjarnsholt, T. Prevalence of Biofilms in Acute Infections Challenges a Longstanding Paradigm. Biofilm 2022, 4, 100080. [Google Scholar] [CrossRef]
- Coenye, T. Biofilm Antimicrobial Susceptibility Testing: Where Are We and Where Could We Be Going? Clin. Microbiol. Rev. 2023, 36, e00024-23. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Nasrollahian, S.; Graham, J.P.; Halaji, M. A Review of the Mechanisms That Confer Antibiotic Resistance in Pathotypes of E. coli. Front. Cell. Infect. Microbiol. 2024, 14, 1387497. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, Y.; Jin, Q.; Ji, J. Emerging Antibacterial Nanomedicine for Enhanced Antibiotic Therapy. Biomater. Sci. 2020, 8, 6825–6839. [Google Scholar] [CrossRef]
- Tian, S.; Van Der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Recent Advances and Future Challenges in the Use of Nanoparticles for the Dispersal of Infectious Biofilms. J. Mater. Sci. Technol. 2021, 84, 208–218. [Google Scholar] [CrossRef]
- Miyata, K.; Christie, R.J.; Kataoka, K. Polymeric Micelles for Nano-Scale Drug Delivery. React. Funct. Polym. 2011, 71, 227–234. [Google Scholar] [CrossRef]
- Toscanini, M.A.; Limeres, M.J.; Garrido, A.V.; Cagel, M.; Bernabeu, E.; Moretton, M.A.; Chiappetta, D.A.; Cuestas, M.L. Polymeric Micelles and Nanomedicines: Shaping the Future of next Generation Therapeutic Strategies for Infectious Diseases. J. Drug Deliv. Sci. Technol. 2021, 66, 102927. [Google Scholar] [CrossRef]
- Ghezzi, M.; Pescina, S.; Padula, C.; Santi, P.; Del Favero, E.; Cantù, L.; Nicoli, S. Polymeric Micelles in Drug Delivery: An Insight of the Techniques for Their Characterization and Assessment in Biorelevant Conditions. J. Control. Release 2021, 332, 312–336. [Google Scholar] [CrossRef]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.-B. Polymeric Micelles as Drug Delivery Vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Xiong, M.-H.; Bao, Y.; Yang, X.-Z.; Zhu, Y.-H.; Wang, J. Delivery of Antibiotics with Polymeric Particles. Adv. Drug Deliv. Rev. 2014, 78, 63–76. [Google Scholar] [CrossRef]
- Chen, J.; Wang, F.; Liu, Q.; Du, J. Antibacterial Polymeric Nanostructures for Biomedical Applications. Chem. Commun. 2014, 50, 14482–14493. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; Wong, E.H.H.; Boyer, C.; Qiao, G.G. Antimicrobial Polymeric Nanoparticles. Progress. Polym. Sci. 2018, 76, 40–64. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; Fernández-García, M. Polymeric Materials with Antimicrobial Activity. Progress. Polym. Sci. 2012, 37, 281–339. [Google Scholar] [CrossRef]
- Borisova, D.; Haladjova, E.; Kyulavska, M.; Petrov, P.; Pispas, S.; Stoitsova, S.; Paunova-Krasteva, T. Application of Cationic Polymer Micelles for the Dispersal of Bacterial Biofilms. Eng. Life Sci. 2018, 18, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Paunova-Krasteva, T.; Haladjova, E.; Petrov, P.; Forys, A.; Trzebicka, B.; Topouzova-Hristova, T.; Stoitsova, S.R. Destruction of Pseudomonas aeruginosa Pre-Formed Biofilms by Cationic Polymer Micelles Bearing Silver Nanoparticles. Biofouling 2020, 36, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In Vitro Cytotoxicity Testing of Polycations: Influence of Polymer Structure on Cell Viability and Hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Knudsen, K.B.; Northeved, H.; Kumar Ek, P.; Permin, A.; Gjetting, T.; Andresen, T.L.; Larsen, S.; Wegener, K.M.; Lykkesfeldt, J.; Jantzen, K.; et al. In Vivo Toxicity of Cationic Micelles and Liposomes. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Stancheva, R.; Paunova-Krasteva, T.; Topouzova-Hristova, T.; Stoitsova, S.; Petrov, P.; Haladjova, E. Ciprofloxacin-Loaded Mixed Polymeric Micelles as Antibiofilm Agents. Pharmaceutics 2023, 15, 1147. [Google Scholar] [CrossRef] [PubMed]
- Kamenova, K.; Prancheva, A.; Radeva, L.; Yoncheva, K.; Zaharieva, M.M.; Najdenski, H.M.; Petrov, P.D. Nanosized Complexes of the Proteolytic Enzyme Serratiopeptidase with Cationic Block Copolymer Micelles Enhance the Proliferation and Migration of Human Cells. Pharmaceutics 2024, 16, 988. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Du, Y.; Xue, Y.; Frisch, H.L. Solubility Parameters. In Physical Properties of Polymers Handbook; Mark, J.E., Ed.; Springer: New York, NY, USA, 2007; pp. 289–303. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Stoitsova, S.; Ivanova, R.; Paunova-Krasteva, T. Biofilm Formation by Reference Strains of Escherichia coli. Comptes Rendus De L’abs 2007, 60, 71–75. [Google Scholar]
- Marhova, M.; Kostadinova, S.; Stoitsova, S. Biofilm-Forming Capabilities of Urinary Escherichia coli Isolates. Biotechnol. Biotechnol. Equip. 2010, 24, 589–593. [Google Scholar] [CrossRef]
- De Soyza, A.; Hall, A.J.; Mahenthiralingam, E.; Drevinek, P.; Kaca, W.; Drulis-Kawa, Z.; Stoitsova, S.R.; Toth, V.; Coenye, T.; Zlosnik, J.E.A.; et al. Developing an International Pseudomonas aeruginosa Reference Panel. MicrobiologyOpen 2013, 2, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.L.; Yang, Q.; Davis, S.; Sampson, E.M.; Azeke, J.I.; Hamad, A.; Schultz, G.S. Antimicrobial Dressing Efficacy against Mature Pseudomonas Aeruginosa Biofilm on Porcine Skin Explants. Int. Wound J. 2015, 12, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Drulis-Kawa, Z. Advantages and Limitations of Microtiter Biofilm Assays in the Model of Antibiofilm Activity of Klebsiella Phage KP34 and Its Depolymerase. Sci. Rep. 2020, 10, 20338. [Google Scholar] [CrossRef] [PubMed]
- De Bleeckere, A.; Van Den Bossche, S.; De Sutter, P.-J.; Beirens, T.; Crabbé, A.; Coenye, T. High Throughput Determination of the Biofilm Prevention Concentration for Pseudomonas Aeruginosa Biofilms Using a Synthetic Cystic Fibrosis Sputum Medium. Biofilm 2023, 5, 100106. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Nguyen, T.H.; Otto, M. The Staphylococcal Exopolysaccharide PIA—Biosynthesis and Role in Biofilm Formation, Colonization, and Infection. Comput. Struct. Biotechnol. J. 2020, 18, 3324–3334. [Google Scholar] [CrossRef] [PubMed]
- Karygianni, L.; Ren, Z.; Koo, H.; Thurnheer, T. Biofilm Matrixome: Extracellular Components in Structured Microbial Communities. Trends Microbiol. 2020, 28, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Balducci, E.; Papi, F.; Capialbi, D.E.; Del Bino, L. Polysaccharides’ Structures and Functions in Biofilm Architecture of Antimicrobial-Resistant (AMR) Pathogens. Int. J. Mol. Sci. 2023, 24, 4030. [Google Scholar] [CrossRef]
- Whitfield, C.; Paiment, A. Biosynthesis and Assembly of Group 1 Capsular Polysaccharides in Escherichia coli and Related Extracellular Polysaccharides in Other Bacteria. Carbohydr. Res. 2003, 338, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Ballén, V.; Cepas, V.; Ratia, C.; Gabasa, Y.; Soto, S.M. Clinical Escherichia coli: From Biofilm Formation to New Antibiofilm Strategies. Microorganisms 2022, 10, 1103. [Google Scholar] [CrossRef]
- Poulin, M.B.; Kuperman, L.L. Regulation of Biofilm Exopolysaccharide Production by Cyclic Di-Guanosine Monophosphate. Front. Microbiol. 2021, 12, 730980. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Huang, J.; Ren, K.; Zhu, Y.; Yang, S. Alginate: Microbial Production, Functionalization, and Biomedical Applications. Int. J. Biol. Macromol. 2023, 242, 125048. [Google Scholar] [CrossRef]
- Franklin, M.J.; Nivens, D.E.; Weadge, J.T.; Howell, P.L. Biosynthesis of the Pseudomonas Aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbio. 2011, 2, e00167. [Google Scholar] [CrossRef]
- Pando, J.M.; Karlinsey, J.E.; Lara, J.C.; Libby, S.J.; Fang, F.C. The Rcs-Regulated Colanic Acid Capsule Maintains Membrane Potential in Salmonella Enterica Serovar Typhimurium. mBio 2017, 8, e00808-17. [Google Scholar] [CrossRef]
- Fulaz, S.; Devlin, H.; Vitale, S.; Quinn, L.; O’Gara, J.P.; Casey, E. Tailoring Nanoparticle-Biofilm Interactions to Increase the Efficacy of Antimicrobial Agents Against Staphylococcus Aureus. IJN 2020, 15, 4779–4791. [Google Scholar] [CrossRef]
- Anastasiadis, P.; Mojica, K.; Layman, C.; Matter, M.L.; Henneman, J.; Barnes, C.; Allen, J.S. Application of Fluorescently Labeled Lectins for the Visualization of Biofilms of Pseudomonas Aeruginosa by High-Frequency Time-Resolved Scanning Acoustic Microscopy. In Proceedings of the 13th International Conference on Biomedical Engineering, IFMBE Proceedings, Singapore, 3–6 December 2008; Lim, C.T., Goh, J.C.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 23, pp. 750–753, ISBN 978-3-540-92840-9. [Google Scholar]
- Vacheva, A.; Ivanova, R.; Paunova-Krasteva, T.; Stoitsova, S. Released Products of Pathogenic Bacteria Stimulate Biofilm Formation by Escherichia coli K-12 Strains. Antonie Van Leeuwenhoek 2012, 102, 105–119. [Google Scholar] [CrossRef]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel Is a Cationic Exopolysaccharide That Cross-Links Extracellular DNA in the Pseudomonas Aeruginosa Biofilm Matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Almuhanna, Y.; Alshahrani, M.Y.; Lowman, D.W.; Rice, P.J.; Gell, C.; Ma, Z.; Graves, B.; Jackson, D.; Lee, K.; et al. Carbohydrates from Pseudomonas Aeruginosa Biofilms Interact with Immune C-Type Lectins and Interfere with Their Receptor Function. Npj Biofilms Microbiomes 2021, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Kromer, C.; Schwibbert, K.; Gadicherla, A.K.; Thiele, D.; Nirmalananthan-Budau, N.; Laux, P.; Resch-Genger, U.; Luch, A.; Tschiche, H.R. Monitoring and Imaging pH in Biofilms Utilizing a Fluorescent Polymeric Nanosensor. Sci. Rep. 2022, 12, 9823. [Google Scholar] [CrossRef] [PubMed]
- Baidya, A.K.; Bhattacharya, S.; Dubey, G.P.; Mamou, G.; Ben-Yehuda, S. Bacterial Nanotubes: A Conduit for Intercellular Molecular Trade. Curr. Opin. Microbiol. 2018, 42, 1–6. [Google Scholar] [CrossRef]
- Dubey, G.P.; Malli Mohan, G.B.; Dubrovsky, A.; Amen, T.; Tsipshtein, S.; Rouvinski, A.; Rosenberg, A.; Kaganovich, D.; Sherman, E.; Medalia, O.; et al. Architecture and Characteristics of Bacterial Nanotubes. Dev. Cell 2016, 36, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on Developments in Synthetic, Analytical, and Medicinal Aspects. J. Enzym. Inhib. Med. Chem. 2010, 25, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Shariati, A.; Arshadi, M.; Khosrojerdi, M.A.; Abedinzadeh, M.; Ganjalishahi, M.; Maleki, A.; Heidary, M.; Khoshnood, S. The Resistance Mechanisms of Bacteria against Ciprofloxacin and New Approaches for Enhancing the Efficacy of This Antibiotic. Front. Public Health 2022, 10, 1025633. [Google Scholar] [CrossRef]
- Aljihani, S.A.; Alehaideb, Z.; Alarfaj, R.E.; Alghoribi, M.F.; Akiel, M.A.; Alenazi, T.H.; Al-Fahad, A.J.; Al Tamimi, S.M.; Albakr, T.M.; Alshehri, A.; et al. Enhancing Azithromycin Antibacterial Activity by Encapsulation in Liposomes/Liposomal-N-Acetylcysteine Formulations Against Resistant Clinical Strains of Escherichia coli. Saudi J. Biol. Sci. 2020, 27, 3065–3071. [Google Scholar] [CrossRef]
- Imperi, F.; Leoni, L.; Visca, P. Antivirulence Activity of Azithromycin in Pseudomonas Aeruginosa. Front. Microbiol. 2014, 5, e00178. [Google Scholar] [CrossRef]
- Imamura, Y.; Higashiyama, Y.; Tomono, K.; Izumikawa, K.; Yanagihara, K.; Ohno, H.; Miyazaki, Y.; Hirakata, Y.; Mizuta, Y.; Kadota, J.; et al. Azithromycin Exhibits Bactericidal Effects on Pseudomonas Aeruginosa Through Interaction with the Outer Membrane. Antimicrob. Agents Chemother. 2005, 49, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Pan, J.; Dong, Y.; Sun, Y.; Wang, Y.; Liao, K.; Chen, Y.; Deng, X.; Yu, S.; Hu, H. Microenvironment Responsive Charge-Switchable Nanoparticles Act on Biofilm Eradication and Virulence Inhibition for Chronic Lung Infection Treatment. J. Control. Release 2024, 365, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Leroy, A.-G.; Caillon, J.; Broquet, A.; Lemabecque, V.; Delanou, S.; Caroff, N.; Asehnoune, K.; Roquilly, A.; Crémet, L. Azithromycin Regulates Bacterial Virulence and Immune Response in a Murine Model of Ceftazidime-Treated Pseudomonas Aeruginosa Acute Pneumonia. Microbiol. Immunol. 2023, 68, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Baidya, A.K.; Rosenshine, I.; Ben-Yehuda, S. Donor-Delivered Cell Wall Hydrolases Facilitate Nanotube Penetration into Recipient Bacteria. Nat. Commun. 2020, 11, 1938. [Google Scholar] [CrossRef]
- Gill, S.; Catchpole, R.; Forterre, P. Extracellular Membrane Vesicles in the Three Domains of Life and Beyond. FEMS Microbiol. Rev. 2019, 43, 273–303. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Baidya, A.K.; Pal, R.R.; Mamou, G.; Gatt, Y.E.; Margalit, H.; Rosenshine, I.; Ben-Yehuda, S. A Ubiquitous Platform for Bacterial Nanotube Biogenesis. Cell Rep. 2019, 27, 334–342.e10. [Google Scholar] [CrossRef]
- Ahmed, F.; Mirani, Z.A.; Ahmed, A.; Urooj, S.; Khan, F.Z.; Siddiqi, A.; Khan, M.N.; Imdad, M.J.; Ullah, A.; Khan, A.B.; et al. Nanotubes Formation in P. aeruginosa. Cells 2022, 11, 3374. [Google Scholar] [CrossRef]
- Sprouse, D.; Jiang, Y.; Laaser, J.E.; Lodge, T.P.; Reineke, T.M. Tuning Cationic Block Copolymer Micelle Size by pH and Ionic Strength. Biomacromolecules 2016, 17, 2849–2859. [Google Scholar] [CrossRef]
- Lorenz, K.; Preem, L.; Sagor, K.; Putrinš, M.; Tenson, T.; Kogermann, K. Development of In Vitro and Ex Vivo Biofilm Models for the Assessment of Antibacterial Fibrous Electrospun Wound Dressings. Mol. Pharm. 2023, 20, 1230–1246. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Yang, Q.; Jeong, S.; Della Porta, A.; Kapoor, H.; Gibson, D.J. A Surfactant-Based Dressing Can Reduce the Appearance of Pseudomonas Aeruginosa Pigments and Uncover the Dermal Extracellular Matrix in an Ex Vivo Porcine Skin Wound Model. Int. Wound J. 2024, 21, e14510. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Alhede, M.; Alhede, M.; Eickhardt-Sørensen, S.R.; Moser, C.; Kühl, M.; Jensen, P.Ø.; Høiby, N. The In Vivo Biofilm. Trends Microbiol. 2013, 21, 466–474. [Google Scholar] [CrossRef]
- Man, M.-Q.; Wakefield, J.S.; Mauro, T.M.; Elias, P.M. Regulatory Role of Nitric Oxide in Cutaneous Inflammation. Inflammation 2022, 45, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Hazell, G.; Khazova, M.; O’Mahoney, P. Low-Dose Daylight Exposure Induces Nitric Oxide Release and Maintains Cell Viability In Vitro. Sci. Rep. 2023, 13, 16306. [Google Scholar] [CrossRef] [PubMed]
- Spernovasilis, N.; Psichogiou, M.; Poulakou, G. Skin Manifestations of Pseudomonas Aeruginosa Infections. Curr. Opin. Infect. Dis. 2021, 32, 72–79. [Google Scholar] [CrossRef]
- Bath, P.M.; Coleman, C.M.; Gordon, A.L.; Lim, W.S.; Webb, A.J. Nitric Oxide for the Prevention and Treatment of Viral, Bacterial, Protozoal and Fungal Infections. F1000Research 2021, 10, 536. [Google Scholar] [CrossRef]
- Ferro, T.A.F.; Souza, E.B.; Suarez, M.A.M.; Rodrigues, J.F.S.; Pereira, D.M.S.; Mendes, S.J.F.; Gonzaga, L.F.; Machado, M.C.A.M.; Bomfim, M.R.Q.; Calixto, J.B.; et al. Topical Application of Cinnamaldehyde Promotes Faster Healing of Skin Wounds Infected with Pseudomonas Aeruginosa. Molecules 2019, 24, 1627. [Google Scholar] [CrossRef] [PubMed]
- Klicznik, M.M.; Szenes-Nagy, A.B.; Campbell, D.J.; Gratz, I.K. Taking the Lead—How Keratinocytes Orchestrate Skin T Cell Immunity. Immunol. Lett. 2018, 200, 43–51. [Google Scholar] [CrossRef]
- Takagi, N.; Kawakami, K.; Kanno, E.; Tanno, H.; Takeda, A.; Ishii, K.; Imai, Y.; Iwakura, Y.; Tachi, M. IL-17A Promotes Neutrophilic Inflammation and Disturbs Acute Wound Healing in Skin. Exp. Dermatol. 2017, 26, 137–144. [Google Scholar] [CrossRef]
- Lecron, J.-C.; Charreau, S.; Jégou, J.-F.; Salhi, N.; Petit-Paris, I.; Guignouard, E.; Burucoa, C.; Favot-Laforge, L.; Bodet, C.; Barra, A.; et al. IL-17 and IL-22 Are Pivotal Cytokines to Delay Wound Healing of S. Aureus and P. Aeruginosa Infected Skin. Front. Immunol. 2022, 13, 984016. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-W.; Chen, Y.-J.; Wang, S.-T.; Ho, L.-W.; Kao, J.-K.; Narita, M.; Takahashi, M.; Wu, C.-Y.; Cheng, H.-Y.; Shieh, J.-J. Azithromycin Impairs TLR7 Signaling in Dendritic Cells and Improves the Severity of Imiquimod-Induced Psoriasis-like Skin Inflammation in Mice. J. Dermatol. Sci. 2016, 84, 59–70. [Google Scholar] [CrossRef]
- Haertlé, J.; Kienlin, P.; Begemann, G.; Werfel, T.; Roesner, L.M. Inhibition of IL-17 Ameliorates Keratinocyte-Borne Cytokine Responses in an In Vitro Model for House-Dust-Mite Triggered Atopic Dermatitis. Sci. Rep. 2023, 13, 16628. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer/Drug | δ [(MPa)1/2] * | χCF | χAZ |
|---|---|---|---|
| PCL | 20.37 | 1.82 | 0.03 |
| PPO | 17.09 | 5.34 | 2.20 |
| CF | 24.87 | - | - |
| AZ | 20.03 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damyanova, T.; Stancheva, R.; Leseva, M.N.; Dimitrova, P.A.; Paunova-Krasteva, T.; Borisova, D.; Kamenova, K.; Petrov, P.D.; Veleva, R.; Zhivkova, I.; et al. Gram Negative Biofilms: Structural and Functional Responses to Destruction by Antibiotic-Loaded Mixed Polymeric Micelles. Microorganisms 2024, 12, 2670. https://doi.org/10.3390/microorganisms12122670
Damyanova T, Stancheva R, Leseva MN, Dimitrova PA, Paunova-Krasteva T, Borisova D, Kamenova K, Petrov PD, Veleva R, Zhivkova I, et al. Gram Negative Biofilms: Structural and Functional Responses to Destruction by Antibiotic-Loaded Mixed Polymeric Micelles. Microorganisms. 2024; 12(12):2670. https://doi.org/10.3390/microorganisms12122670
Chicago/Turabian StyleDamyanova, Tsvetozara, Rumena Stancheva, Milena N. Leseva, Petya A. Dimitrova, Tsvetelina Paunova-Krasteva, Dayana Borisova, Katya Kamenova, Petar D. Petrov, Ralitsa Veleva, Ivelina Zhivkova, and et al. 2024. "Gram Negative Biofilms: Structural and Functional Responses to Destruction by Antibiotic-Loaded Mixed Polymeric Micelles" Microorganisms 12, no. 12: 2670. https://doi.org/10.3390/microorganisms12122670
APA StyleDamyanova, T., Stancheva, R., Leseva, M. N., Dimitrova, P. A., Paunova-Krasteva, T., Borisova, D., Kamenova, K., Petrov, P. D., Veleva, R., Zhivkova, I., Topouzova-Hristova, T., Haladjova, E., & Stoitsova, S. (2024). Gram Negative Biofilms: Structural and Functional Responses to Destruction by Antibiotic-Loaded Mixed Polymeric Micelles. Microorganisms, 12(12), 2670. https://doi.org/10.3390/microorganisms12122670