
Investigation of Staphylococcus aureus Biofilm-Associated Toxin as a Potential Squamous Cell Carcinoma Therapeutic


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Bacterial Strains
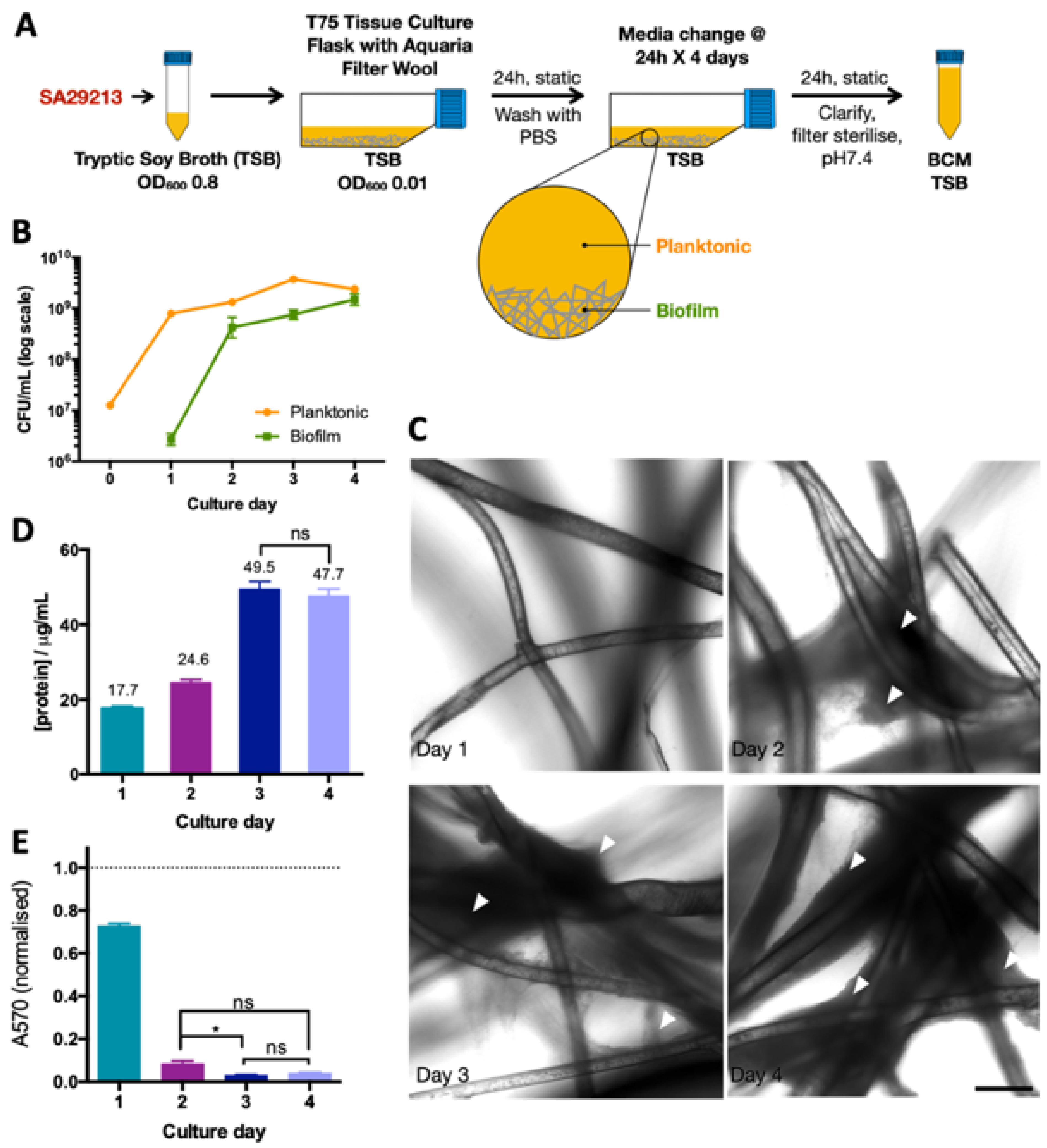
2.3. Biofilm-Conditioned Media Preparation
2.4. BCM Inactivation
2.5. BCM Fractionation by Fast Protein Liquid Chromatography (FLPC)
2.6. SCC-12/HaCaT Co-Culture
2.7. Live/Dead Fluorescence Microscopy
2.8. Cell Viability and Caspase Activity Assay
2.9. Immunocytochemistry
2.10. Western Blot
2.11. Transposon Mutagenesis
2.12. Reverse-Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.13. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) and Protein Identification (ID)
3. Results
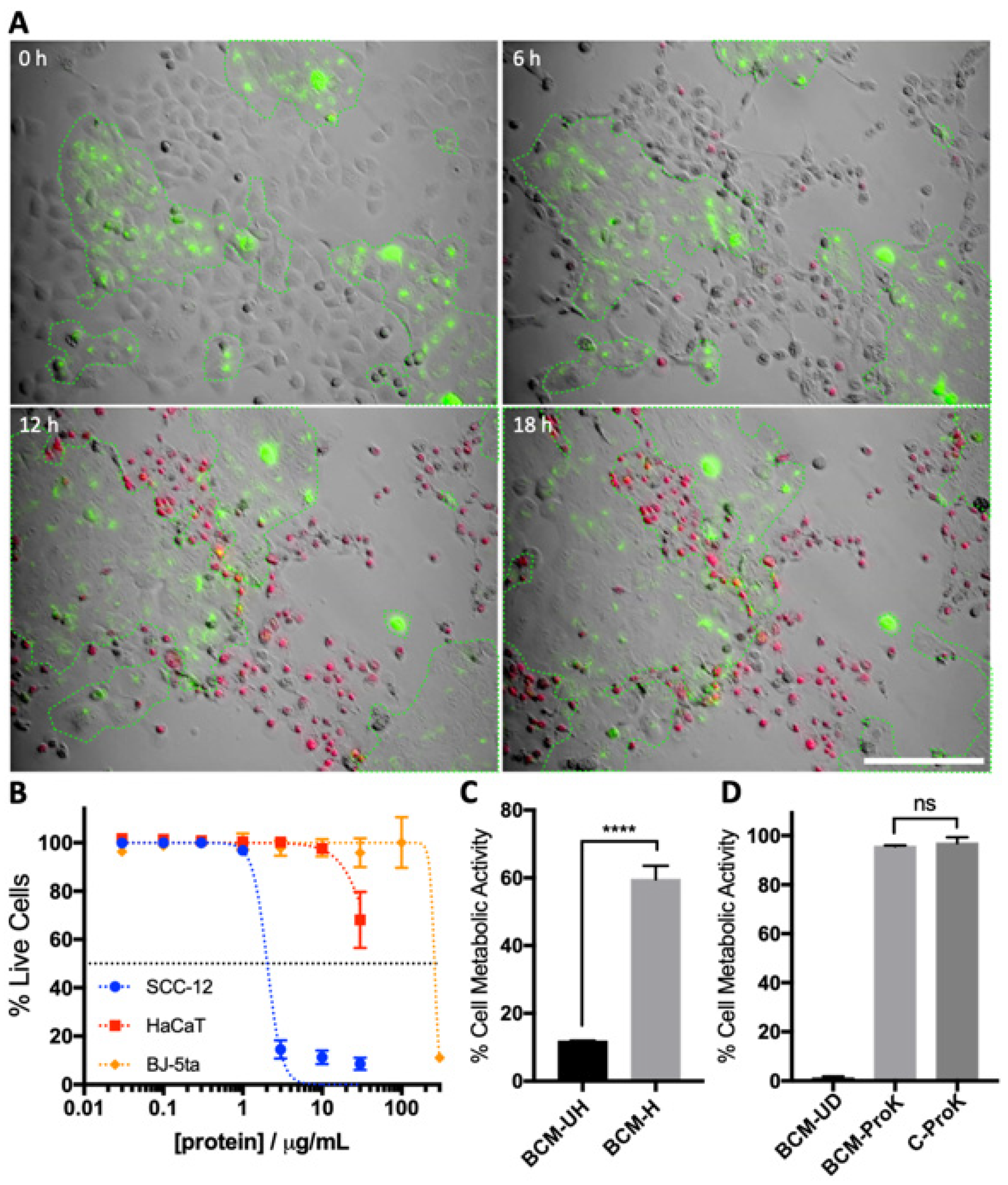
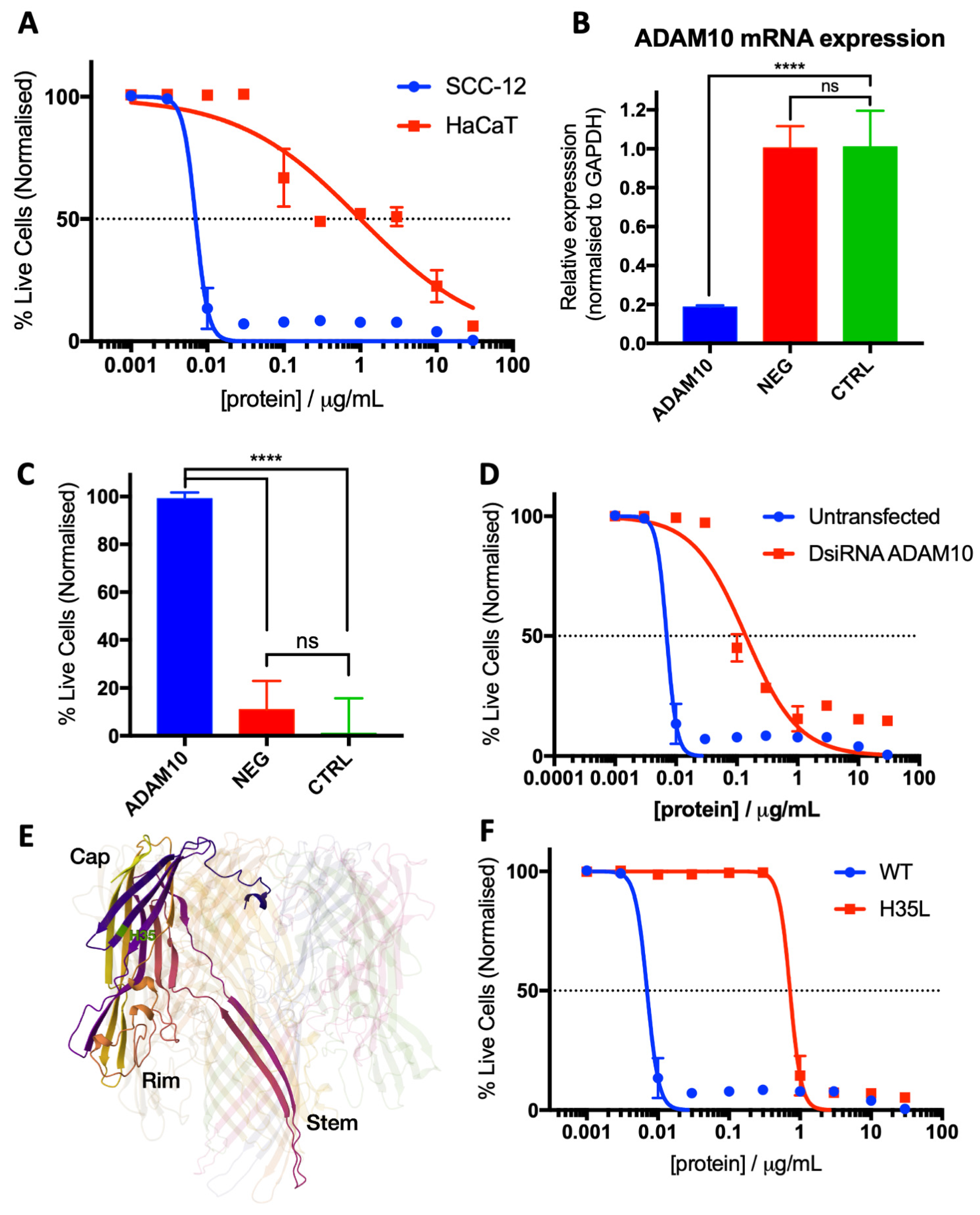
3.1. S. aureus BCM Displays Selective Toxicity towards Cancerous Keratinocytes
3.2. Characterisation of SA29213 BCM Toxicity on Cancerous and Non-Cancerous Keratinocytes
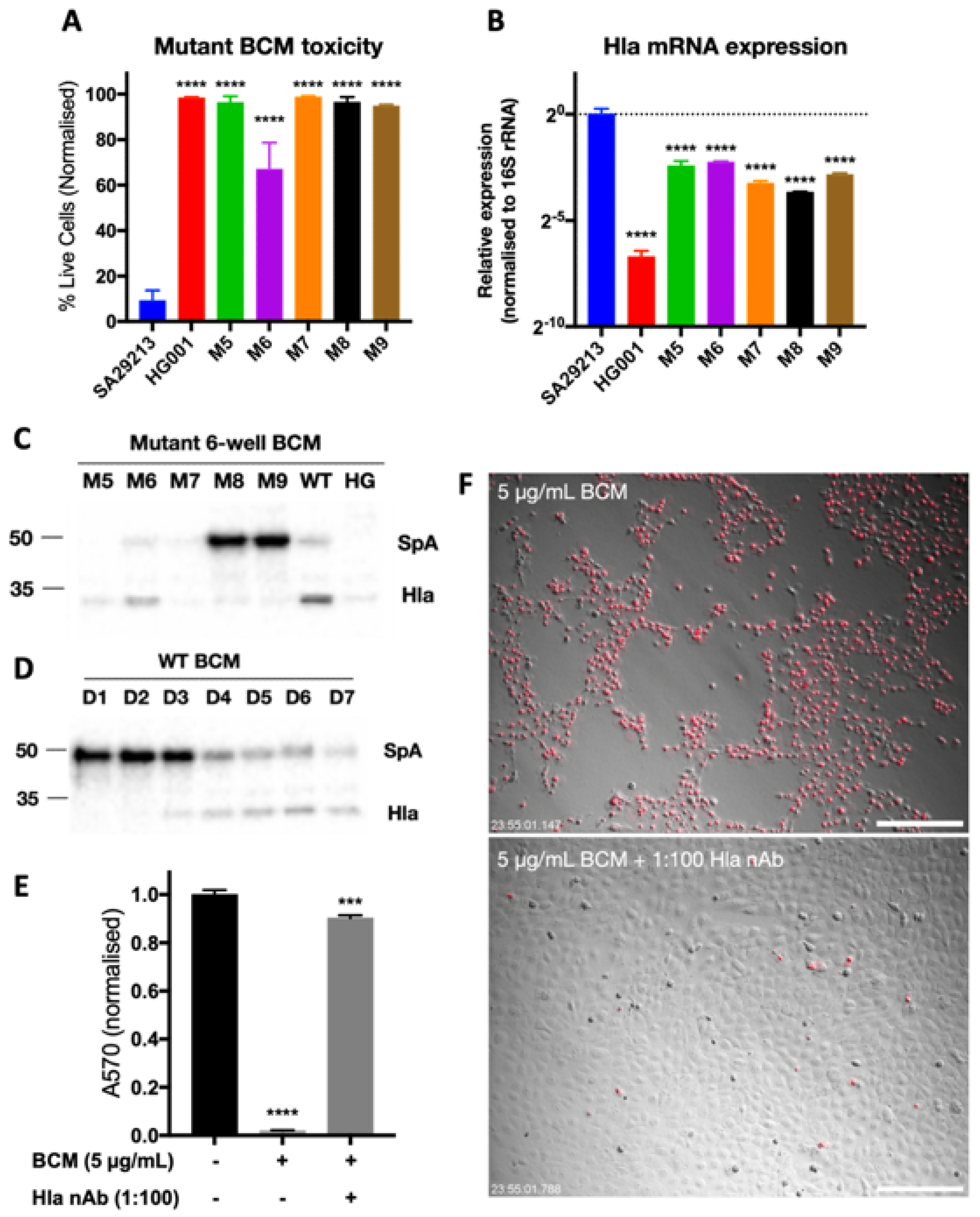
3.3. Identification of SCC-12-Specific SA29213 Protein Toxin
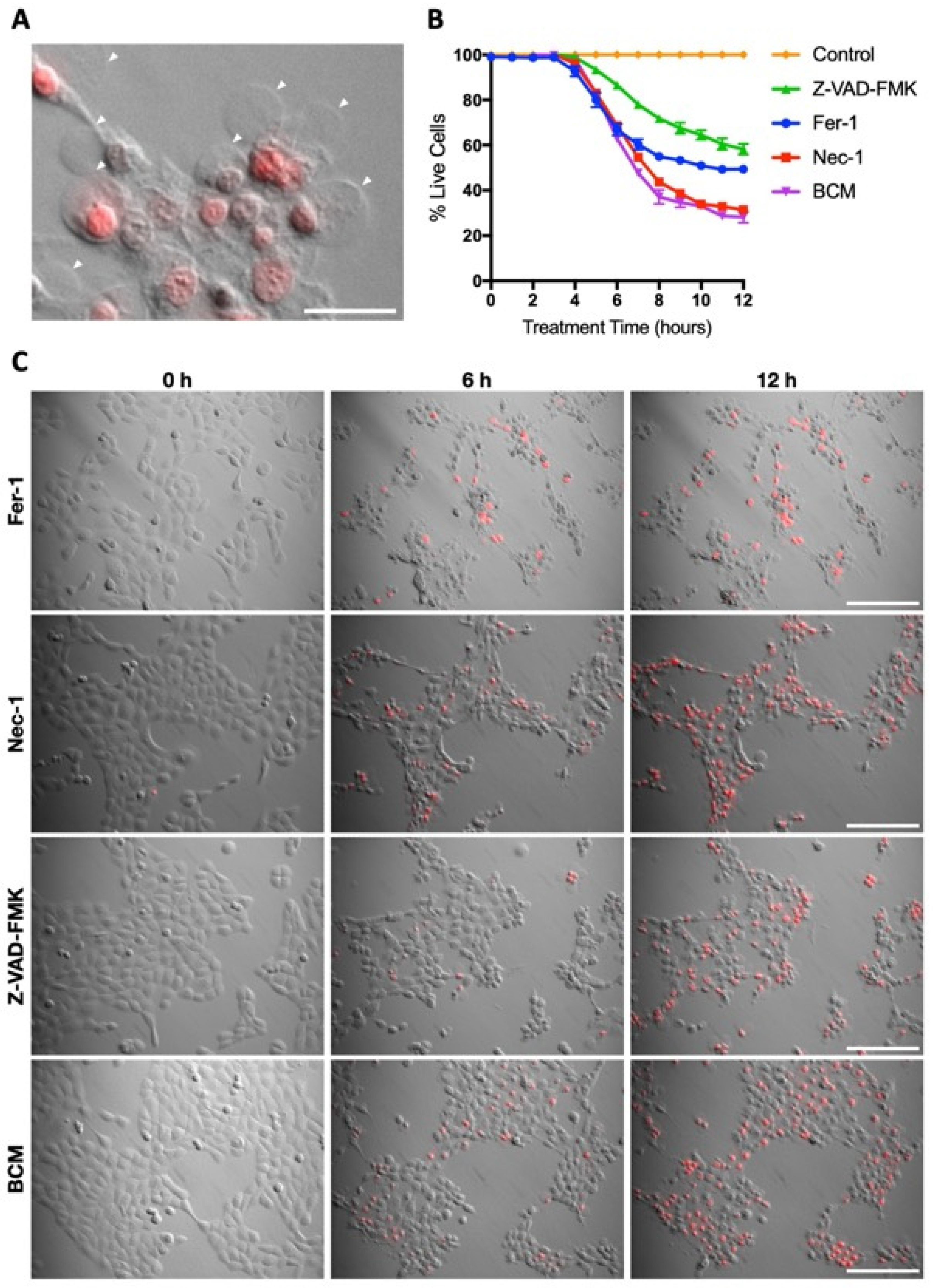
3.4. Characterisation of Hla as an SCC-12-Specific Protein Toxin
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stratigos, A.; Garbe, C.; Lebbe, C.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Becker, J.C.; Zalaudek, I.; Saiag, P.; et al. Diagnosis and treatment of invasive squamous cell carcinoma of the skin: European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 1989–2007. [Google Scholar] [CrossRef]
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.C.; Jin, A.; Koh, W.P. Trends of cutaneous basal cell carcinoma, squamous cell carcinoma, and melanoma among the Chinese, Malays, and Indians in Singapore from 1968-2016. JAAD Int. 2021, 4, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.C.; Federman, D.G.; Kirsner, R.S. Skin cancer as an occupational disease: The effect of ultraviolet and other forms of radiation. Int. J. Dermatol. 2005, 44, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Clayman, G.L.; Lee, J.J.; Holsinger, F.C.; Zhou, X.; Duvic, M.; El-Naggar, A.K.; Prieto, V.G.; Altamirano, E.; Tucker, S.L.; Strom, S.S.; et al. Mortality risk from squamous cell skin cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Armstrong, B.K.; Kricker, A. The epidemiology of UV induced skin cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Wolff, K.; Johnson, R.A.; Saavedra, A.P. Precancerous Lesions and Cutaneous Carcinomas. In Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 7th ed.; McGraw-Hill Education: New York, NY, USA, 2016. [Google Scholar]
- Lindelof, B.; Sigurgeirsson, B.; Gabel, H.; Stern, R.S. Incidence of skin cancer in 5356 patients following organ transplantation. Br. J. Dermatol. 2000, 143, 513–519. [Google Scholar]
- Mudigonda, T.; Levender, M.M.; O’Neill, J.L.; West, C.E.; Pearce, D.J.; Feldman, S.R. Incidence, risk factors, and preventative management of skin cancers in organ transplant recipients: A review of single- and multicenter retrospective studies from 2006 to 2010. Dermatol. Surg. Off. Publ. Am. Soc. Dermatol. Surg. 2013, 39, 345–364. [Google Scholar] [CrossRef]
- Zhao, H.; Shu, G.; Wang, S. The risk of non-melanoma skin cancer in HIV-infected patients: New data and meta-analysis. Int. J. STD AIDS 2016, 27, 568–575. [Google Scholar] [CrossRef]
- Swanson, N.A. Mohs surgery. Technique, indications, applications, and the future. Arch. Dermatol. 1983, 119, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Leibovitch, I.; Huilgol, S.C.; Selva, D.; Hill, D.; Richards, S.; Paver, R. Cutaneous squamous cell carcinoma treated with Mohs micrographic surgery in Australia I. Experience over 10 years. J. Am. Acad. Dermatol. 2005, 53, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.E.; Carroll, R.J.; Day, C.L., Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J. Am. Acad. Dermatol. 1992, 26, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Imiquimod Cream. Available online: http://www.cancerresearchuk.org/about-cancer/skin-cancer/treatment/imiquimod-cream (accessed on 19 February 2018).
- British Association of Dermatologists. Imiquimod Creams. Available online: http://www.bad.org.uk/shared/get-file.ashx?id=209&itemtype=document (accessed on 23 February 2023).
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. IJMM 2009, 299, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Bhave, M.S.; Hassanbhai, A.M.; Anand, P.; Luo, K.Q.; Teoh, S.H. Effect of Heat-Inactivated Clostridium sporogenes and Its Conditioned Media on 3-Dimensional Colorectal Cancer Cell Models. Sci. Rep. 2015, 5, 15681. [Google Scholar] [CrossRef] [PubMed]
- Ansiaux, R.; Baudelet, C.; Cron, G.O.; Segers, J.; Dessy, C.; Martinive, P.; De Wever, J.; Verrax, J.; Wauthier, V.; Beghein, N.; et al. Botulinum toxin potentiates cancer radiotherapy and chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1276–1283. [Google Scholar] [CrossRef]
- Johansson, D.; Bergstrom, P.; Henriksson, R.; Grankvist, K.; Johansson, A.; Behnam-Motlagh, P. Adenylate cyclase toxin from Bordetella pertussis enhances cisplatin-induced apoptosis to lung cancer cells in vitro. Oncol. Res. 2006, 15, 423–430. [Google Scholar] [CrossRef]
- Frankel, A.E.; Powell, B.L.; Lilly, M.B. Diphtheria toxin conjugate therapy of cancer. Cancer Chemother. Biol. Response Modif. 2002, 20, 301–313. [Google Scholar]
- Frankel, A.E.; Kreitman, R.J.; Sausville, E.A. Targeted toxins. Clin. Cancer Res. 2000, 6, 326–334. [Google Scholar]
- Nakatsuji, T.; Chen, T.H.; Butcher, A.M.; Trzoss, L.L.; Nam, S.-J.; Shirakawa, K.T.; Zhou, W.; Oh, J.; Otto, M.; Fenical, W.; et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci. Adv. 2018, 4, eaao4502. [Google Scholar] [CrossRef]
- Jenkins, A.; Diep, B.A.; Mai, T.T.; Vo, N.H.; Warrener, P.; Suzich, J.; Stover, C.K.; Sellman, B.R. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio 2015, 6, e02272-14. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Castleman, M.J.; Febbraio, M.; Hall, P.R. CD36 Is Essential for Regulation of the Host Innate Response to Staphylococcus aureus α-Toxin-Mediated Dermonecrosis. J. Immunol. 2015, 195, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- McElroy, M.C.; Harty, H.R.; Hosford, G.E.; Boylan, G.M.; Pittet, J.F.; Foster, T.J. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 1999, 67, 5541–5544. [Google Scholar] [CrossRef] [PubMed]
- Seeger, W.; Birkemeyer, R.G.; Ermert, L.; Suttorp, N.; Bhakdi, S.; Duncker, H.R. Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab. Investig. 1990, 63, 341–349. [Google Scholar] [PubMed]
- Seeger, W.; Bauer, M.; Bhakdi, S. Staphylococcal alpha-toxin elicits hypertension in isolated rabbit lungs. Evidence for thromboxane formation and the role of extracellular calcium. J. Clin. Investig. 1984, 74, 849–858. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Bae, T.; Otto, M.; Deleo, F.R.; Schneewind, O. Poring over pores: Alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 2007, 13, 1405–1406. [Google Scholar] [CrossRef]
- Callegan, M.C.; Engel, L.S.; Hill, J.M.; O’Callaghan, R.J. Corneal virulence of Staphylococcus aureus: Roles of alpha-toxin and protein A in pathogenesis. Infect. Immun. 1994, 62, 2478–2482. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, R.J.; Callegan, M.C.; Moreau, J.M.; Green, L.C.; Foster, T.J.; Hartford, O.M.; Engel, L.S.; Hill, J.M. Specific roles of alpha-toxin and beta-toxin during Staphylococcus aureus corneal infection. Infect. Immun. 1997, 65, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.E.; Kim, H.K.; Wang, Y.; Bubeck Wardenburg, J. ADAM10 mediates vascular injury induced by Staphylococcus aureus α-hemolysin. J. Infect. Dis. 2012, 206, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Menzies, B.E.; Kernodle, D.S. Passive immunization with antiserum to a nontoxic alpha-toxin mutant from Staphylococcus aureus is protective in a murine model. Infect. Immun. 1996, 64, 1839–1841. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Bae, T.; Glass, E.M.; Schneewind, O.; Missiakas, D. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 2008, 416, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Soldani, C.; Lazzè, M.C.; Bottone, M.G.; Tognon, G.; Biggiogera, M.; Pellicciari, C.E.; Scovassi, A.I. Poly(ADP-ribose) polymerase cleavage during apoptosis: When and where? Exp. Cell Res. 2001, 269, 193–201. [Google Scholar] [CrossRef]
- Bae, T.; Banger, A.K.; Wallace, A.; Glass, E.M.; Aslund, F.; Schneewind, O.; Missiakas, D.M. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 2004, 101, 12312–12317. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Bayley, H. Interactions between residues in staphylococcal alpha-hemolysin revealed by reversion mutagenesis. J. Biol. Chem. 1995, 270, 23072–23076. [Google Scholar] [CrossRef]
- Jursch, R.; Hildebrand, A.; Hobom, G.; Tranum-Jensen, J.; Ward, R.; Kehoe, M.; Bhakdi, S. Histidine residues near the N terminus of staphylococcal alpha-toxin as reporters of regions that are critical for oligomerization and pore formation. Infect. Immun. 1994, 62, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yan, M.; Ji, Y. The H35A mutated alpha-toxin interferes with cytotoxicity of staphylococcal alpha-toxin. Infect. Immun. 2009, 77, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Menzies, B.E.; Kernodle, D.S. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: Role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 1994, 62, 1843–1847. [Google Scholar] [CrossRef] [PubMed]
- Krishnasastry, M.; Walker, B.; Braha, O.; Bayley, H. Surface labeling of key residues during assembly of the transmembrane pore formed by staphylococcal alpha-hemolysin. FEBS Lett. 1994, 356, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Dragneva, Y.; Anuradha, C.D.; Valeva, A.; Hoffmann, A.; Bhakdi, S.; Husmann, M. Subcytocidal attack by staphylococcal alpha-toxin activates NF-kappaB and induces interleukin-8 production. Infect. Immun. 2001, 69, 2630–2635. [Google Scholar] [CrossRef]
- Giese, B.; Dittmann, S.; Paprotka, K.; Levin, K.; Weltrowski, A.; Biehler, D.; Lâm, T.-T.; Sinha, B.; Fraunholz, M.J. Staphylococcal alpha-toxin is not sufficient to mediate escape from phagolysosomes in upper-airway epithelial cells. Infect. Immun. 2009, 77, 3611–3625. [Google Scholar] [CrossRef]
- Kirker, K.R.; James, G.A.; Fleckman, P.; Olerud, J.E.; Stewart, P.S. Differential effects of planktonic and biofilm MRSA on human fibroblasts. Wound Repair. Regen. 2012, 20, 253–261. [Google Scholar] [CrossRef]
- Tankersley, A.; Frank, M.B.; Bebak, M.; Brennan, R. Early effects of Staphylococcus aureus biofilm secreted products on inflammatory responses of human epithelial keratinocytes. J. Inflamm. 2014, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.J., Jr.; Ward, C.L.; Romano, D.R.; Hurtgen, B.J.; Hardy, S.K.; Woodbury, R.L.; Trevino, A.V.; Rathbone, C.R.; Wenke, J.C. Staphylococcus aureus biofilms decrease osteoblast viability, inhibits osteogenic differentiation, and increases bone resorption in vitro. BMC Musculoskelet. Disord. 2013, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.L.; Sanchez, C.J., Jr.; Pollot, B.E.; Romano, D.R.; Hardy, S.K.; Becerra, S.C.; Rathbone, C.R.; Wenke, J.C. Soluble factors from biofilms of wound pathogens modulate human bone marrow-derived stromal cell differentiation, migration, angiogenesis, and cytokine secretion. BMC Microbiol. 2015, 15, 75. [Google Scholar] [CrossRef] [PubMed]
- Secor, P.R.; James, G.A.; Fleckman, P.; Olerud, J.E.; McInnerney, K.; Stewart, P.S. Staphylococcus aureus Biofilm and Planktonic cultures differentially impact gene expression, mapk phosphorylation, and cytokine production in human keratinocytes. BMC Microbiol. 2011, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.C.; Leonard, A.; Schäuble, M.; Rieckmann, L.M.; Hoyer, J.; Maass, S.; Lalk, M.; Becher, D.; Pané-Farré, J.; Riedel, K. Virulence Factors Produced by Staphylococcus aureus Biofilms Have a Moonlighting Function Contributing to Biofilm Integrity. Mol. Cell Proteom. 2019, 18, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Marano, R.J.; Wallace, H.J.; Wijeratne, D.; Fear, M.W.; San Wong, H.; O’Handley, R. Secreted biofilm factors adversely affect cellular wound healing responses in vitro. Sci. Rep. 2015, 5, 13296. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.S.; Núñez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Ezekwe, E.A.D., Jr.; Weng, C.; Duncan, J.A. ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus α-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins 2016, 8, 95. [Google Scholar] [CrossRef]
- St Jean, A.T.; Swofford, C.A.; Panteli, J.T.; Brentzel, Z.J.; Forbes, N.S. Bacterial delivery of Staphylococcus aureus alpha-hemolysin causes regression and necrosis in murine tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1266–1274. [Google Scholar] [CrossRef]
- Swofford, C.A.; St Jean, A.T.; Panteli, J.T.; Brentzel, Z.J.; Forbes, N.S. Identification of Staphylococcus aureus alpha-hemolysin as a protein drug that is secreted by anticancer bacteria and rapidly kills cancer cells. Biotechnol. Bioeng. 2014, 111, 1233–1245. [Google Scholar] [CrossRef]
- Johansson, D.; Johansson, A.; Behnam-Motlagh, P. alpha-Toxin of Staphylococcus aureus overcomes acquired cisplatin-resistance in malignant mesothelioma cells. Cancer Lett. 2008, 265, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Cheley, S.; Bayley, H. Redirecting Pore Assembly of Staphylococcal α-Hemolysin by Protein Engineering. ACS Cent. Sci. 2019, 5, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Yamashita, D.; Kato, K.; Peng, Z.; Ueda, J.; Kaneko, J.; Kamio, Y.; Tanaka, Y.; Yao, M. Structural basis for pore-forming mechanism of staphylococcal α-hemolysin. Toxicon 2015, 108, 226–231. [Google Scholar] [CrossRef]
- Valeva, A.; Pongs, J.; Bhakdi, S.; Palmer, M. Staphylococcal alpha-toxin: The role of the N-terminus in formation of the heptameric pore -- a fluorescence study. Biochim. Biophys. Acta 1997, 1325, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Palmer, M.; Bhakdi, S. Staphylococcal alpha-toxin: Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages. Biochemistry 1997, 36, 13298–13304. [Google Scholar] [CrossRef]
- Walker, B.; Krishnasastry, M.; Zorn, L.; Bayley, H. Assembly of the oligomeric membrane pore formed by Staphylococcal alpha-hemolysin examined by truncation mutagenesis. J. Biol. Chem. 1992, 267, 21782–21786. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Weisser, A.; Walker, B.; Kehoe, M.; Bayley, H.; Bhakdi, S.; Palmer, M. Molecular architecture of a toxin pore: A 15-residue sequence lines the transmembrane channel of staphylococcal alpha-toxin. EMBO J. 1996, 15, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Bayley, H. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 1995, 270, 23065–23071. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Crawford, H.C.; Dempsey, P.J.; Brown, G.; Adam, L.; Moss, M.L. ADAM10 as a therapeutic target for cancer and inflammation. Curr. Pharm. Des. 2009, 15, 2288–2299. [Google Scholar] [CrossRef]
- Smith, T.M., Jr.; Tharakan, A.; Martin, R.K. Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, K.; Sakai, K.; Hongo, K.; Fukushima, M.; Kawakubo, M.; Nakayama, J. High expression of ADAM10 predicts a poor prognosis for patients with glioblastoma. Int. J. Clin. Exp. Pathol. 2017, 10, 618–624. [Google Scholar]
- Zheng, H.; Zhong, A.; Xie, S.; Wang, Y.; Sun, J.; Zhang, J.; Tong, Y.; Chen, M.; Zhang, G.; Ma, Q.; et al. Elevated serum HER-2 predicts poor prognosis in breast cancer and is correlated to ADAM10 expression. Cancer Med. 2019, 8, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Zhang, P.; Hou, Q.; Feng, S.; Liu, L.; Cui, D.; Shi, H.; Fu, Y.; Luo, Y. A novel pan-cancer biomarker plasma heat shock protein 90alpha and its diagnosis determinants in clinic. Cancer Sci. 2019, 110, 2941–2959. [Google Scholar] [CrossRef] [PubMed]
- Stasikowska-Kanicka, O.; Wągrowska-Danilewicz, M.; Kulicka, P.; Danilewicz, M. Overexpression of ADAM10 in oral squamous cell carcinoma with metastases. Pol. J. Pathol. 2018, 69, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Lambert, D.W.; Speight, P.M.; Whawell, S.A. ADAM 10 is over expressed in oral squamous cell carcinoma and contributes to invasive behaviour through a functional association with αvβ6 integrin. FEBS Lett. 2013, 587, 3529–3534. [Google Scholar] [CrossRef]
- Kai, Y.; Peng, W.; Ling, W.; Jiebing, H.; Zhuan, B. Reciprocal effects between microRNA-140-5p and ADAM10 suppress migration and invasion of human tongue cancer cells. Biochem. Biophys. Res. Commun. 2014, 448, 308–314. [Google Scholar] [CrossRef]
- Guo, J.; He, L.; Yuan, P.; Wang, P.; Lu, Y.; Tong, F.; Wang, Y.; Yin, Y.; Tian, J.; Sun, J. ADAM10 overexpression in human non-small cell lung cancer correlates with cell migration and invasion through the activation of the Notch1 signaling pathway. Oncol. Rep. 2012, 28, 1709–1718. [Google Scholar] [CrossRef]
- Oh, S.T.; Stark, A.; Reichrath, J. The disintegrin-metalloproteinases ADAM10 and ADAM17 are upregulated in cutaneous squamous cell carcinomas. Derm. Endocrinol. 2016, 8, e1228499. [Google Scholar] [CrossRef]
- Becker, R.E.N.; Berube, B.J.; Sampedro, G.R.; DeDent, A.C.; Bubeck Wardenburg, J. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus α-toxin. J. Innate Immun. 2014, 6, 619–631. [Google Scholar] [CrossRef]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Salemi, J.; Hou, H.; Zhu, Y.; Mori, T.; Giunta, B.; Obregon, D.; Tan, J. Rapamycin promotes beta-amyloid production via ADAM-10 inhibition. Biochem. Biophys. Res. Commun. 2010, 398, 337–341. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, Z.X.; Kannan, B.; Phillips, A.R.J.; Becker, D.L. Investigation of Staphylococcus aureus Biofilm-Associated Toxin as a Potential Squamous Cell Carcinoma Therapeutic. Microorganisms 2024, 12, 293. https://doi.org/10.3390/microorganisms12020293
Ong ZX, Kannan B, Phillips ARJ, Becker DL. Investigation of Staphylococcus aureus Biofilm-Associated Toxin as a Potential Squamous Cell Carcinoma Therapeutic. Microorganisms. 2024; 12(2):293. https://doi.org/10.3390/microorganisms12020293
Chicago/Turabian StyleOng, Zi Xin, Bavani Kannan, Anthony R. J. Phillips, and David L. Becker. 2024. "Investigation of Staphylococcus aureus Biofilm-Associated Toxin as a Potential Squamous Cell Carcinoma Therapeutic" Microorganisms 12, no. 2: 293. https://doi.org/10.3390/microorganisms12020293