


Improved Canker Processing and Viability Droplet Digital PCR Allow Detection of Erwinia amylovora Viable Nonculturable Cells in Apple Bark



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain, Culture Media, and Inoculum Preparation
2.2. Plant Material Collection and Cryopreservation
2.3. Plant Material Processing Optimization
2.4. PMAxx Dye Treatment and DNA Extraction
2.5. Comparison of Different Commercial and Manual DNA Extraction Methods
2.6. Transfer of a Chip-Based dCPR to a ddPCR Platform
2.7. Correlation between ddPCR Data and Plate Counts
2.8. Selective Detection of Viable E. amylovora Cells Using PMAxx and the QX200 ddPCR System
2.9. Analysis of E. amylovora Total, Live, and Culturable Cell Populations in Fire Blight Cankers and the Peripheric Symptomless Bark Areas
2.10. Statistical Analysis
3. Results and Discussion
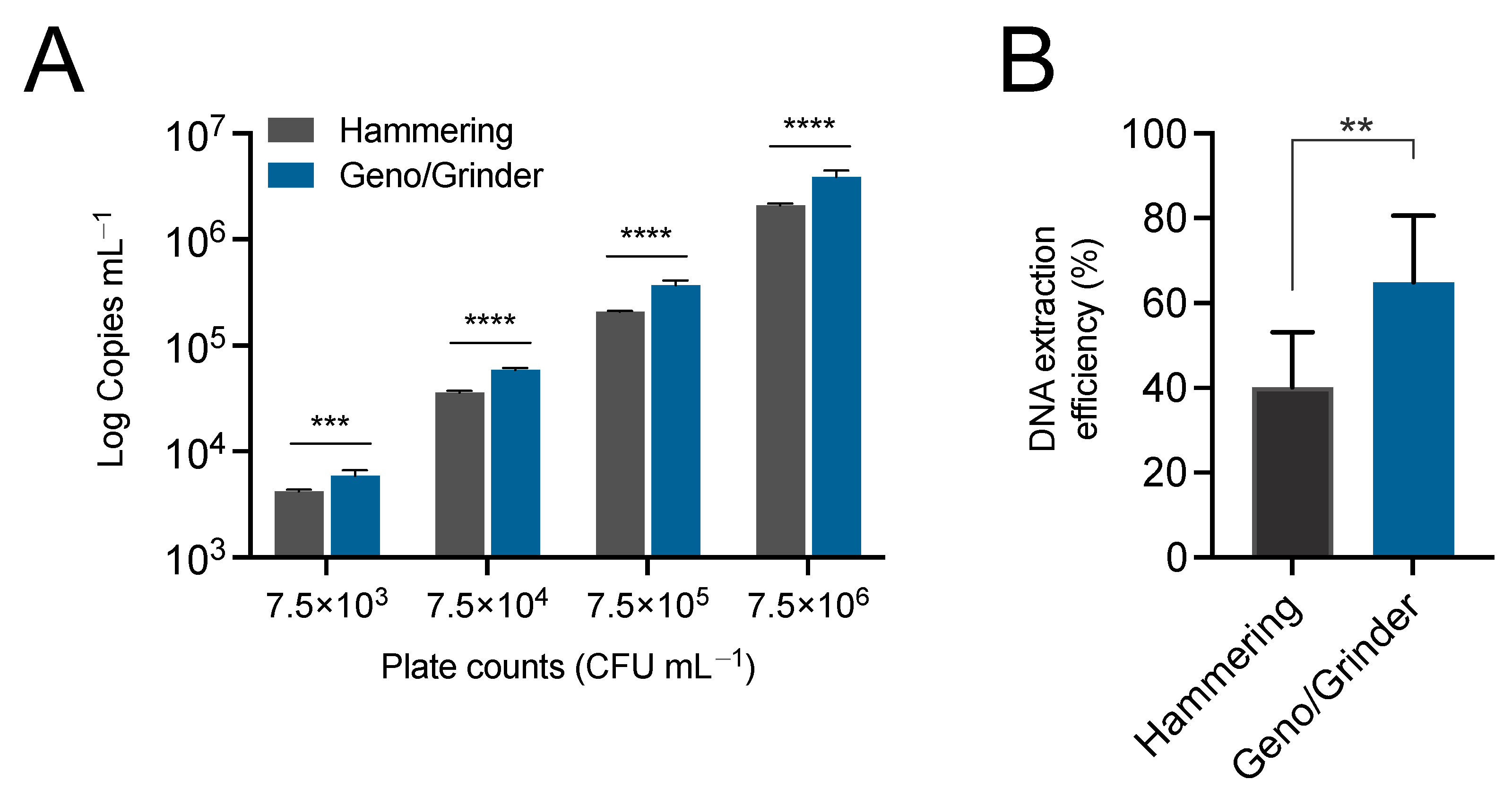
3.1. Automated Canker Homogenization Improves Recovery of E. amylovora from Cankers
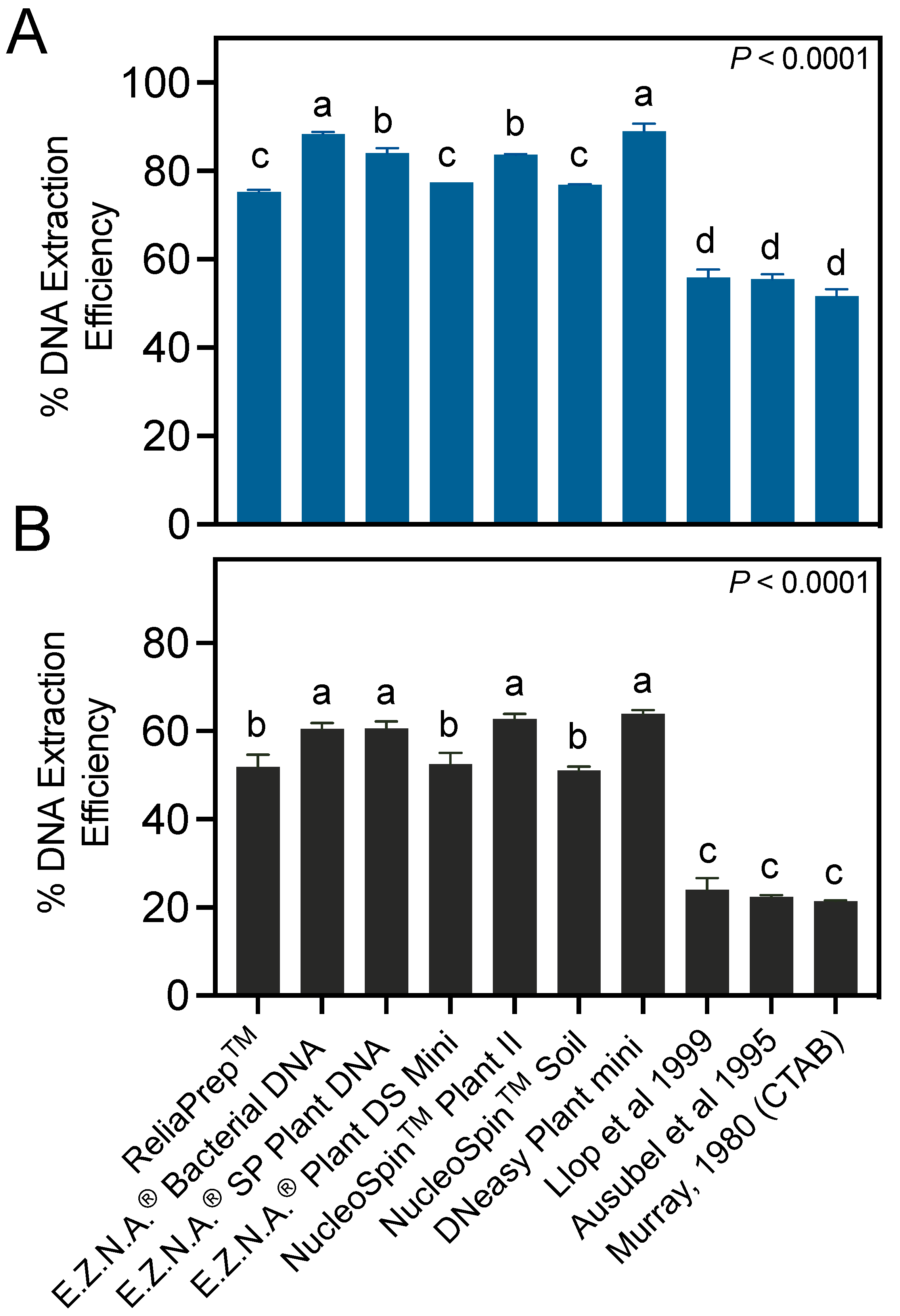
3.2. Column-Based Commercial Kits Provide Higher DNA Extraction Efficiencies than Manual Protocols
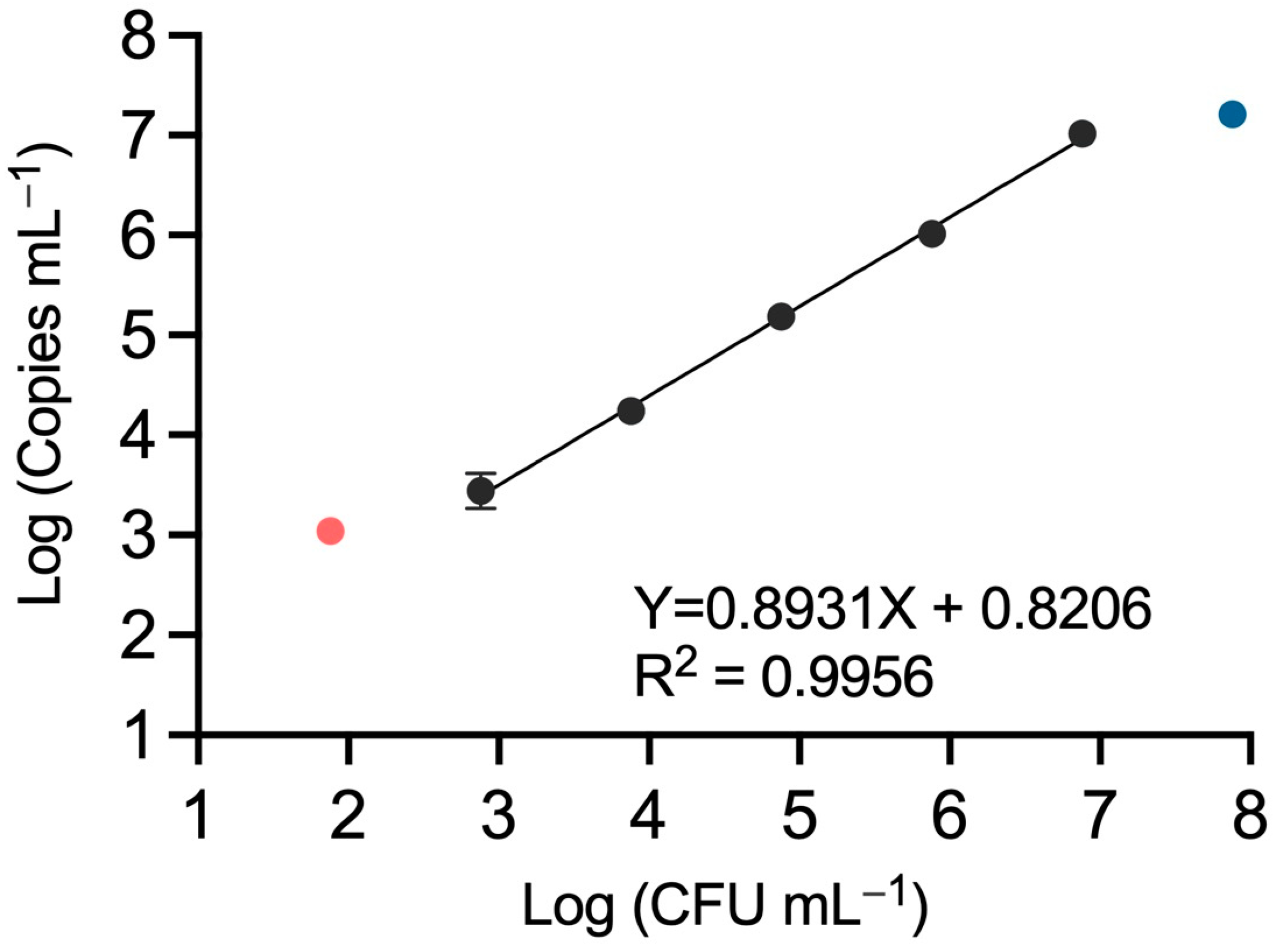
3.3. QX200 Droplet dPCR Provides Similar Quantification Dynamic Range Compared to the Chip-Based QS3D dPCR
3.4. The Transferred v-ddPCR Protocol Efficiently Discriminates between Live and Dead E. amylovora Cells
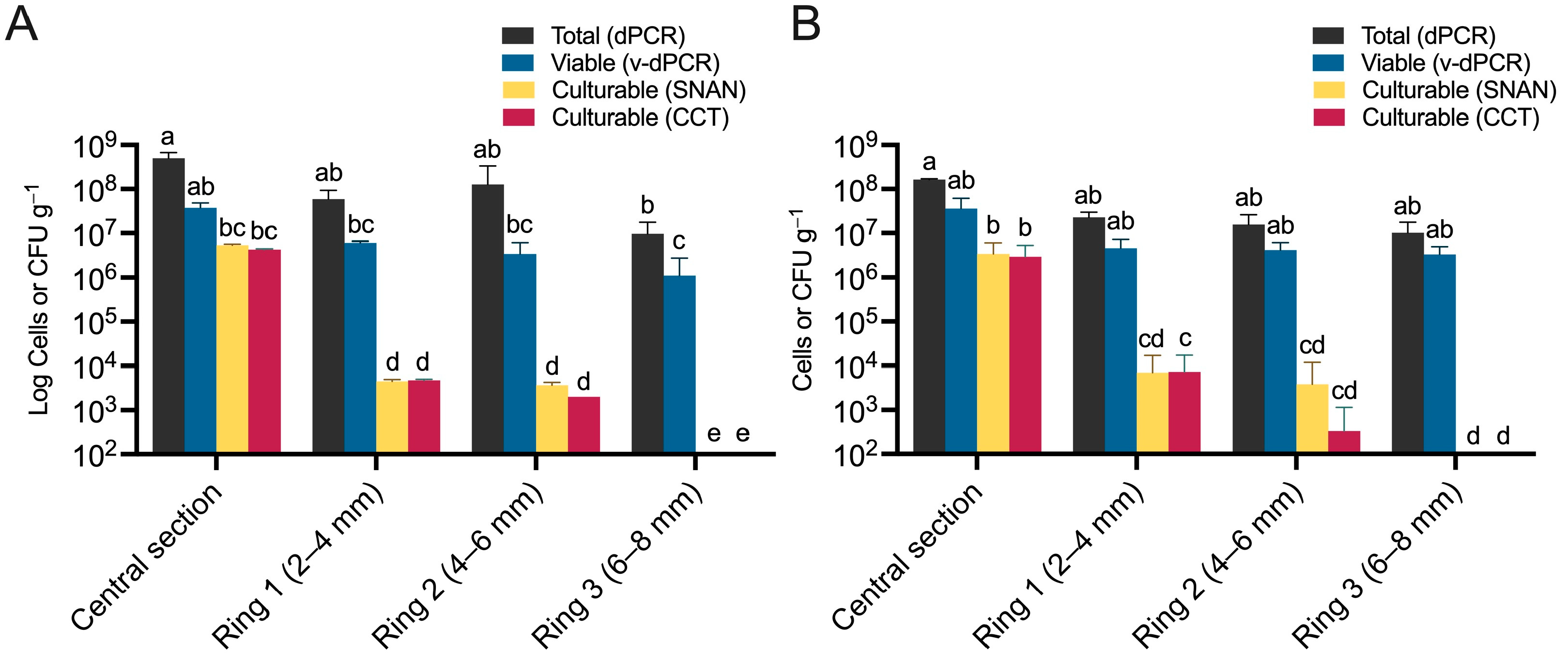
3.5. Viable E. amylovora Populations in Tissues within and around Cankers Are Mostly Composed of VBNC Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Zwet, T.; Orolaza-Halbrendt, N.; Zeller, W. Fire Blight History, Biology, and Management; American Phytopathological Society: St. Paul, MN, USA, 2016. [Google Scholar]
- Vanneste, J. Erwinia amylovora (fireblight). In CABI Compendium.; CAB International: Wallingford, UK, 2023. [Google Scholar] [CrossRef]
- Thomson, S.V. Epidemiology of fire blight. In Fire Blight: The Disease and Its Causative Agent, Erwinia amylovora; Vanneste, J.L., Ed.; CAB International: Wallingford, UK, 2000; pp. 9–36. [Google Scholar]
- Aćimović, S.G.; Santander, R.D.; Meredith, C.L.; Pavlović, Ž.M. Fire blight rootstock infections causing apple tree death: A case study in high-density apple orchards with Erwinia amylovora strain characterization. Front. Hortic. 2023, 2, 1082204. [Google Scholar] [CrossRef]
- Santander, R.D.; Meredith, C.L.; Aćimović, S.G. Development of a viability digital PCR protocol for the selective detection and quantification of live Erwinia amylovora cells in cankers. Sci. Rep. 2019, 9, 11530. [Google Scholar] [CrossRef] [PubMed]
- Santander, R.D.; Gašić, K.; Aćimović, S.G. Selective quantification of Erwinia amylovora live cells in pome fruit tree cankers by viability digital PCR. Methods Mol. Biol. 2022, 2536, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Nocker, A.; Cheung, C.-Y.; Camper, A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 2006, 67, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Nocker, A.; Sossa-Fernandez, P.; Burr, M.D.; Camper, A.K. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl. Environ. Microbiol. 2007, 73, 5111–5117. [Google Scholar] [CrossRef] [PubMed]
- Santander, R.D.; Khodadadi, F.; Meredith, C.L.; Rađenović, Ž.; Clements, J.; Aćimović, S.G. Fire blight resistance, irrigation and conducive wet weather improve Erwinia amylovora winter survival in cankers. Front. Microbiol. 2022, 13, 1009364. [Google Scholar] [CrossRef] [PubMed]
- Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar] [CrossRef]
- Dye, D.W. A taxonomic study of the genus Erwinia. I. The ‘amylocora’ group. N. Z. J. Sci. 1968, 11, 590–607. [Google Scholar]
- Pedersen, J.C. Natamycin as a fungicide in agar media. Appl. Environ. Microbiol. 1992, 58, 1064–1066. [Google Scholar] [CrossRef]
- Ishimaru, C.; Klos, E.J. New medium for detecting Erwinia amylovora and its use in epidemiological studies. Phytopathology 1984, 74, 1342–1345. [Google Scholar] [CrossRef]
- Anonymous. EPPO standard on diagnostics: PM 7/20 (3) Erwinia amylovora. EPPO Bull. 2022, 52, 198–224. [Google Scholar] [CrossRef]
- Llop, P.; Caruso, P.; Cubero, J.; Morente, C.; López, M.M. A simple extraction procedure for efficient routine detection of pathogenic bacteria in plant material by polymerase chain reaction. J. Microbiol. Methods 1999, 37, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, P. Short Protocols in Molecular Biology: A Compendium of Methods From “Current Protocols in Molecular Biology”, 2nd ed.; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1992; Volume 275, pp. 28764–28773. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- Pirc, M.; Ravnikar, M.; Tomlinson, J.; Dreo, T. Improved fireblight diagnostics using quantitative real-time PCR detection of Erwinia amylovora chromosomal DNA. Plant Pathol. 2009, 58, 872–881. [Google Scholar] [CrossRef]
- Pavšič, J.; Žel, J.; Milavec, M. Assessment of the real-time PCR and different digital PCR platforms for DNA quantification. Anal. Bioanal. Chem. 2016, 408, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Demeke, T.; Jenkins, G.R. Influence of DNA Extraction Methods, PCR Inhibitors and Quantification Methods on Real-Time PCR Assay of Biotechnology-Derived Traits. Anal. Bioanal. Chem. 2010, 396, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Rohde, A.; Hammerl, J.A.; Appel, B.; Dieckmann, R.; Al Dahouk, S. Sampling and Homogenization Strategies Significantly Influence the Detection of Foodborne Pathogens in Meat. Biomed Res. Int. 2015, 2015, 145437. [Google Scholar] [CrossRef]
- Holden, M.J.; Blasic, J.R.; Bussjaeger, L.; Kao, C.; Shokere, L.A.; Kendall, D.C.; Freese, L.; Jenkins, G.R. Evaluation of extraction methodologies for corn kernel (Zea mays) DNA for detection of trace amounts of biotechnology-derived DNA. J. Agric. Food Chem. 2003, 51, 2468–2474. [Google Scholar] [CrossRef]
- Moreano, F.; Busch, U.; Engel, K.-H. Distortion of genetically modified organism quantification in processed foods: Influence of particle size compositions and heat-induced DNA degradation. J. Agric. Food Chem. 2005, 53, 9971–9979. [Google Scholar] [CrossRef]
- Aboul-Maaty, N.A.-F.; Oraby, H.A.-S. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. Natl. Res. Cent. 2019, 43, 25. [Google Scholar] [CrossRef]
- Heikrujam, J.; Kishor, R.; Behari Mazumder, P. The chemistry behind plant DNA isolation protocols. In Biochemical Analysis Tools—Methods for Bio-Molecules Studies; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar]
- Rucińska, A.; Olszak, M.; Świerszcz, S.; Nobis, M.; Zubek, S.; Kusza, G.; Boczkowska, M.; Nowak, A. Looking for hidden enemies of metabarcoding: Species composition, habitat and management can strongly influence dna extraction while examining grassland communities. Biomolecules 2021, 11, 318. [Google Scholar] [CrossRef]
- Bahnweg, G.; Schulze, S.; Möller, E.M.; Rosenbrock, H.; Langebartels, C.; Sandermann, H. DNA isolation from recalcitrant materials such as tree roots, bark, and forest soil for the detection of fungal pathogens by polymerase chain reaction. Anal. Biochem. 1998, 262, 79–82. [Google Scholar] [CrossRef]
- Moreira, P.A.; Oliveira, D.A. Leaf age affects the quality of DNA extracted from Dimorphandra mollis (Fabaceae), a tropical tree species from the cerrado region of brazil. Genet. Mol. Res. 2011, 10, 353–358. [Google Scholar] [CrossRef]
- Claassen, S.; du Toit, E.; Kaba, M.; Moodley, C.; Zar, H.J.; Nicol, M.P. A Comparison of the efficiency of five different commercial DNA extraction kits for extraction of DNA from faecal samples. J. Microbiol. Methods 2013, 94, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kemp, B.M.; Winters, M.; Monroe, C.; Barta, J.L. How much DNA is lost? Measuring DNA loss of short-tandem-repeat length fragments targeted by the PowerPlex 16® system using the Qiagen MinElute Purification Kit. Hum. Biol. 2014, 86, 313. [Google Scholar] [CrossRef]
- Taylor, R.K.; Guilford, P.J.; Clark, R.G.; Hale, C.N.; Forster, R.L.S. Detection of Erwinia amylovora in plant material using novel polymerase chain reaction (PCR) primers. N. Z. J. Crop Hortic. Sci. 2001, 29, 35–43. [Google Scholar] [CrossRef]
- Dreo, T.; Pirc, M.; Ramšak, Ž.; Pavšič, J.; Milavec, M.; Žel, J.; Gruden, K. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: A case study of fire blight and potato brown rot. Anal. Bioanal. Chem. 2014, 406, 6513–6528. [Google Scholar] [CrossRef]
- Morisset, D.; Štebih, D.; Milavec, M.; Gruden, K.; Žel, J. Quantitative analysis of food and feed samples with droplet digital PCR. PLoS ONE 2013, 8, e62583. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.M.; Busby, E.; Garson, J.A.; Grant, P.R.; Nastouli, E.; Devonshire, A.S.; Whale, A.S. Digital PCR dynamic range is approaching that of real-time quantitative PCR. Biomol. Detect. Quantif. 2016, 10, 31–33. [Google Scholar] [CrossRef]
- Li, J.; Zhai, S.; Gao, H.; Xiao, F.; Li, Y.; Wu, G.; Wu, Y. Development and assessment of a duplex droplet digital PCR method for quantification of GM rice Kemingdao. Anal. Bioanal. Chem. 2021, 413, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, P.; Lu, L.; Zhong, H.; Xu, M.; Jia, R.; Su, L.; Cao, L.; Sun, Y.; Guo, M.; et al. Development of a multiplex droplet digital PCR assay for detection of enterovirus, parechovirus, herpes simplex virus 1 and 2 simultaneously for diagnosis of viral CNS infections. Virol. J. 2022, 19, 70. [Google Scholar] [CrossRef]
- He, Z.; Wang, J.; Fike, B.J.; Li, X.; Li, C.; Mendis, B.L.; Li, P. A portable droplet generation system for ultra-wide dynamic range digital PCR based on a vibrating sharp-tip capillary. Biosens. Bioelectron. 2021, 191, 113458. [Google Scholar] [CrossRef]
- Shum, E.Y.; Lai, J.H.; Li, S.; Lee, H.G.; Soliman, J.; Raol, V.K.; Lee, C.K.; Fodor, S.P.A.; Fan, H.C. Next-generation digital polymerase chain reaction: High-dynamic-range single-molecule DNA counting via ultrapartitioning. Anal. Chem. 2022, 94, 17868–17876. [Google Scholar] [CrossRef]
- Fittipaldi, M.; Nocker, A.; Codony, F. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J. Microbiol. Methods 2012, 91, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Gao, Y.; Miya, S.; Kuda, T.; Kimura, B. discrimination of live and dead cells of Escherichia coli using propidium monoazide after sodium dodecyl sulfate treatment. Food Control 2017, 71, 79–82. [Google Scholar] [CrossRef]
- Papanicolas, L.E.; Wang, Y.; Choo, J.M.; Gordon, D.L.; Wesselingh, S.L.; Rogers, G.B. optimisation of a propidium monoazide based method to determine the viability of microbes in faecal slurries for transplantation. J. Microbiol. Methods 2019, 156, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Codony, F.; Dinh-Thanh, M.; Agustí, G. key factors for removing bias in viability PCR-based methods: A review. Curr. Microbiol. 2020, 77, 682–687. [Google Scholar] [CrossRef]
- Beer, S.; Norelli, J. Fire blight epidemiology: Factors affecting release of Erwinia amylovora by cankers. Phytopathology 1977, 67, 1119–1125. [Google Scholar] [CrossRef]
- Biggs, A.R. Characteristics of fire blight cankers following shoot inoculations of three apple cultivars. Acta Hortic. 1994, 29, 795–797. [Google Scholar] [CrossRef]
- Kielak, K.; Sobiczewski, P.; Pulawska, J. Overwintering of Erwinia amylovora in naturally and artificially infected appie shoots. Acta Hortic. 2002, 590, 157–162. [Google Scholar] [CrossRef]
- Ordax, M.; Marco-Noales, E.; López, M.M.; Biosca, E.G. Survival strategy of Erwinia amylovora against copper: Induction of the viable-but-nonculturable state. Appl. Environ. Microbiol. 2006, 72, 3482–3488. [Google Scholar] [CrossRef]
- Ordax, M.; Biosca, E.G.; Wimalajeewa, S.C.; López, M.M.; Marco-Noales, E. Survival of Erwinia amylovora in mature apple fruit calyces through the viable but nonculturable (VBNC) state. J. Appl. Microbiol. 2009, 107, 106–116. [Google Scholar] [CrossRef]
- Biosca, E.G.; Santander, R.D.; Ordax, M.; Marco-Noales, E.; Águila, B.; Flores, A.; López, M.M. Survival of Erwinia amylovora in rain water at low temperatures. In Current Research Topics in Applied Microbiology and Microbial Biotechnology; Mendez-Vilas, A., Ed.; World Scientific Publishing: Singapore, 2009; pp. 88–91. [Google Scholar] [CrossRef]
- Santander, R.D.; Catalá-Senent, J.F.; Ordax, M.; Flores, A.; Marco-Noales, E.; Biosca, E.G. Evaluation of flow cytometry to assess Erwinia amylovora viability under different stress conditions. In Microorganisms in Industry and Environment; Mendez-Vilas, A., Ed.; World Scientific Publishing: Singapore, 2010; pp. 51–54. [Google Scholar] [CrossRef]
- Santander, R.D.; Català-Senent, J.F.; Marco-Noales, E.; Biosca, E.G. in planta recovery of Erwinia amylovora viable but nonculturable cells. Trees 2012, 26, 75–82. [Google Scholar] [CrossRef]
- Santander, R.D.; Oliver, J.D.; Biosca, E.G. cellular, physiological, and molecular adaptive responses of Erwinia amylovora to starvation. FEMS Microbiol. Ecol. 2014, 88, 258–271. [Google Scholar] [CrossRef]
- Santander, R.D.; Biosca, E.G. Erwinia amylovora psychrotrophic adaptations: Evidence of pathogenic potential and survival at temperate and low environmental temperatures. PeerJ 2017, 5, e3931. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, Y.J.; Park, D.H. extended longevity of Erwinia amylovora vectored by honeybees under in vitro conditions and its capacity for dissemination. Plant Pathol. 2022, 71, 762–771. [Google Scholar] [CrossRef]
- Barcina, I.; Arana, I. The viable but nonculturable phenotype: A crossroads in the life-cycle of non-differentiating bacteria? Rev. Environ. Sci. Biotechnol. 2009, 8, 245–255. [Google Scholar] [CrossRef]
- Oliver, J.D. recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol. Rev. 2010, 34, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Santos, M.A.; Chambel, L. thirty years of viable but nonculturable state research: Unsolved molecular mechanisms. Crit. Rev. Microbiol. 2015, 41, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Sobiczewski, P.; Pulawska, J.; Berczyński, S.; Konicka, M. Fire blight detection and control in Poland. Acta Hortic. 1999, 489, 115–120. [Google Scholar] [CrossRef]
- Sobiczewski, P.; Kielak, K.; Puławska, J.; Berczynski, S. Winter survival of Erwinia amylovora in apple terminal shoot tissue. Phytopathol. Pol. 2006, 39, 93–103. [Google Scholar]
- Sobiczewski, P.; Iakimova, E.T.; Mikiciński, A.; Węgrzynowicz-Lesiak, E.; Dyki, B. Necrotrophic behaviour of Erwinia amylovora in apple and tobacco leaf tissue. Plant Pathol. 2017, 66, 842–855. [Google Scholar] [CrossRef]
- Ordax, M.; Piquer-Salcedo, J.E.; Santander, R.D.; Sabater-Muñoz, B.; Biosca, E.G.; López, M.M.; Marco-Noales, E. Medfly Ceratitis capitata as potential vector for fire blight pathogen Erwinia amylovora: Survival and transmission. PLoS ONE 2015, 10, e0127560. [Google Scholar] [CrossRef] [PubMed]
- Grey, B.E.; Steck, T.R. the viable but nonculturable state of Ralstonia solanacearum may be involved in long-term survival and plant infection. Appl. Environ. Microbiol. 2001, 67, 3866–3872. [Google Scholar] [CrossRef]
- Mock, N.M.; Baker, C.J.; Aver’yanov, A.A. induction of a viable but not culturable (VBNC) state in some Pseudomonas syringae pathovars upon exposure to oxidation of an apoplastic phenolic, acetosyringone. Physiol. Mol. Plant Pathol. 2015, 89, 16–24. [Google Scholar] [CrossRef]
- Szatmári, Á.; Móricz, Á.M.; Schwarczinger, I.; Kolozsváriné Nagy, J.; Alberti, Á.; Pogány, M.; Bozsó, Z. A pattern-triggered immunity-related phenolic, acetosyringone, boosts rapid inhibition of a diverse set of plant pathogenic bacteria. BMC Plant Biol. 2021, 21, 153. [Google Scholar] [CrossRef] [PubMed]
- Venisse, J.-S.; Gullner, G.; Brisset, M.-N. evidence for the involvement of an oxidative stress in the initiation of infection of pear by Erwinia amylovora. Plant Physiol. 2001, 125, 2164–2172. [Google Scholar] [CrossRef]
- Venisse, J.-S.; Barny, M.-A.; Paulin, J.-P.; Brisset, M.-N. Involvement of three pathogenicity factors of Erwinia amylovora in the oxidative stress associated with compatible interaction in pear. FEBS Lett. 2003, 537, 198–202. [Google Scholar] [CrossRef]
- Santander, R.D.; Monte-Serrano, M.; Rodríguez-Herva, J.J.; López-Solanilla, E.; Rodríguez-Palenzuela, P.; Biosca, E.G. exploring new roles for the rpoS gene in the survival and virulence of the fire blight pathogen Erwinia amylovora. FEMS Microbiol. Ecol. 2014, 90, 895–907. [Google Scholar] [CrossRef]
- Santander, R.D.; Figàs-Segura, À.; Biosca, E.G. Erwinia amylovora catalases KatA and KatG are virulence factors and delay the starvation-induced viable but non-culturable (VBNC) response. Mol. Plant Pathol. 2018, 19, 922–934. [Google Scholar] [CrossRef]
- Morcia, C.; Ghizzoni, R.; Delogu, C.; Andreani, L.; Carnevali, P.; Terzi, V. Digital PCR: What relevance to plant studies? Biology 2020, 9, 433. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CFU mL−1 | Replicate | Copies L−1 | CV% a | Copies mL−1 b | Positive Calls | Accepted Calls |
|---|---|---|---|---|---|---|
| 7.60 × 107 | 1 | 9992.19 | 13.10 | 1.67 × 107 | 13,822 | 13,825 |
| 2 | 10,983.49 | 1.83 × 107 | 16,408 | 16,410 | ||
| 3 | 8435.36 | 1.41 × 107 | 12,502 | 12,505 | ||
| 7.60 × 106 | 1 | 5996.34 | 4.02 | 9.99 × 105 | 16,835 | 16,897 |
| 2 | 6485.90 | 1.08 × 106 | 16,789 | 16,857 | ||
| 3 | 6345.67 | 1.06 × 106 | 16,342 | 16,645 | ||
| 6.60 × 105 | 1 | 546.99 | 10.83 | 9.12 × 105 | 5744 | 11,596 |
| 2 | 679.84 | 1.13 × 106 | 5352 | 12,194 | ||
| 3 | 627.34 | 1.05 × 106 | 5281 | 11,093 | ||
| 7.60 ×104 | 1 | 87.05 | 5.55 | 1.45 × 105 | 725 | 9245 |
| 2 | 97.14 | 1.62 × 105 | 736 | 9287 | ||
| 3 | 93.78 | 1.56 × 105 | 729 | 9164 | ||
| 7.60 × 103 | 1 | 10.12 | 9.61 | 1.69 × 104 | 84 | 10,039 |
| 2 | 11.60 | 1.93 × 104 | 100 | 10,195 | ||
| 3 | 9.68 | 1.61 × 104 | 76 | 10,119 | ||
| 7.60 × 102 | 1 | 2.06 | 20.44 | 3.43 × 103 | 12 | 13,666 |
| 2 | 2.12 | 3.53 × 103 | 13 | 13,678 | ||
| 3 | 1.43 | 2.23 × 103 | 11 | 13,665 | ||
| 7.60 × 101 | 1 | 0.72 | 13.21 | 1.20 × 103 | 4 | 16,375 |
| 2 | 0.56 | 9.33 × 102 | 7 | 10,257 | ||
| 3 | 0.7 | 1.17 × 103 | 5 | 13,065 | ||
| NTC c,d | 1 | 1.59 | 108.26 | 2.17 × 102 | 7 | 14,902 |
| 2 | 0.26 | 1.67 × 102 | 3 | 14,484 | ||
| 3 | 0.27 | 1.83 × 102 | 4 | 14,327 | ||
| NTC c | 1 | 0 | 173.21 | 0.00 | 0 | 13,127 |
| 2 | 0 | 0.00 | 0 | 9385 | ||
| 3 | 0.21 | 2.00 | 2 | 11,291 | ||
| NTC c | 1 | 0 | 87.05 | 0.00 | 0 | 14,902 |
| 2 | 0.08 | 1.00 | 1 | 14,484 | ||
| 3 | 0.09 | 1.00 | 1 | 14,327 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhar, B.C.; Delgado Santander, R.; Aćimović, S.G. Improved Canker Processing and Viability Droplet Digital PCR Allow Detection of Erwinia amylovora Viable Nonculturable Cells in Apple Bark. Microorganisms 2024, 12, 376. https://doi.org/10.3390/microorganisms12020376
Dhar BC, Delgado Santander R, Aćimović SG. Improved Canker Processing and Viability Droplet Digital PCR Allow Detection of Erwinia amylovora Viable Nonculturable Cells in Apple Bark. Microorganisms. 2024; 12(2):376. https://doi.org/10.3390/microorganisms12020376
Chicago/Turabian StyleDhar, Bidhan Chandra, Ricardo Delgado Santander, and Srđan G. Aćimović. 2024. "Improved Canker Processing and Viability Droplet Digital PCR Allow Detection of Erwinia amylovora Viable Nonculturable Cells in Apple Bark" Microorganisms 12, no. 2: 376. https://doi.org/10.3390/microorganisms12020376
APA StyleDhar, B. C., Delgado Santander, R., & Aćimović, S. G. (2024). Improved Canker Processing and Viability Droplet Digital PCR Allow Detection of Erwinia amylovora Viable Nonculturable Cells in Apple Bark. Microorganisms, 12(2), 376. https://doi.org/10.3390/microorganisms12020376