
Extraction Methods Determine the Quality of Soil Microbiota Acquisition
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling
2.2. Design of the Soil Microbiota Extraction Methods
2.3. Investigation of the Effect Size of Sonication Time and Strategy among the Extracted Microbiota
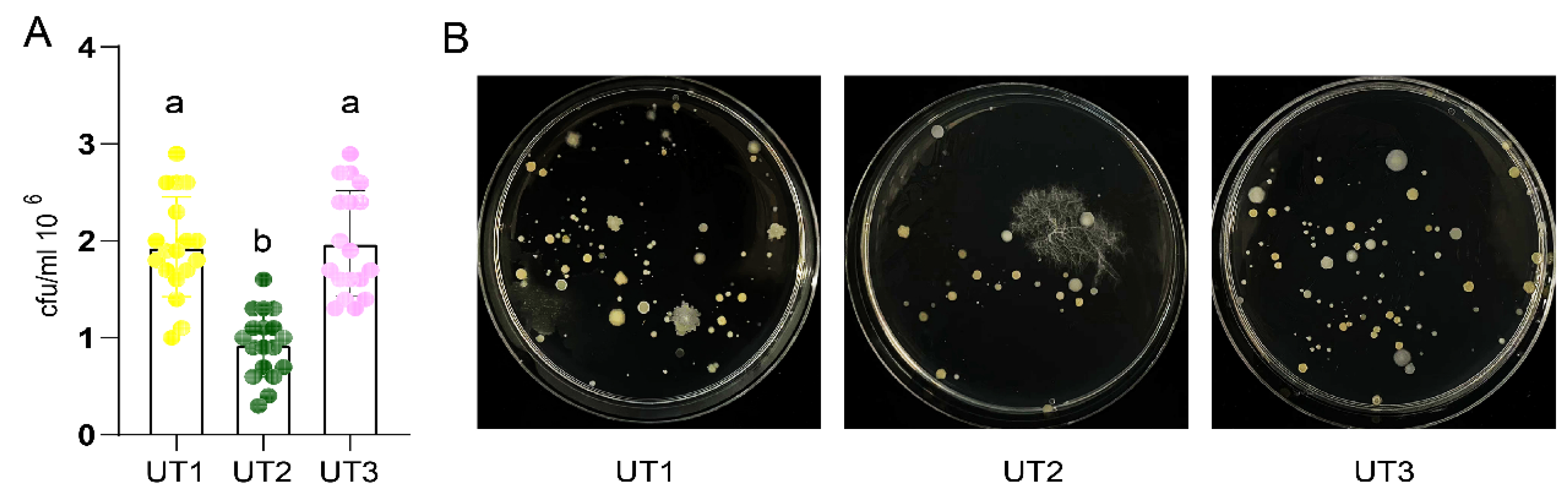
2.4. Plate Counting
2.5. 16s rRNA Amplicon Sequencing
2.6. Data Analysis
3. Results
3.1. Sonication Increased Culturable Bacteria Diversity
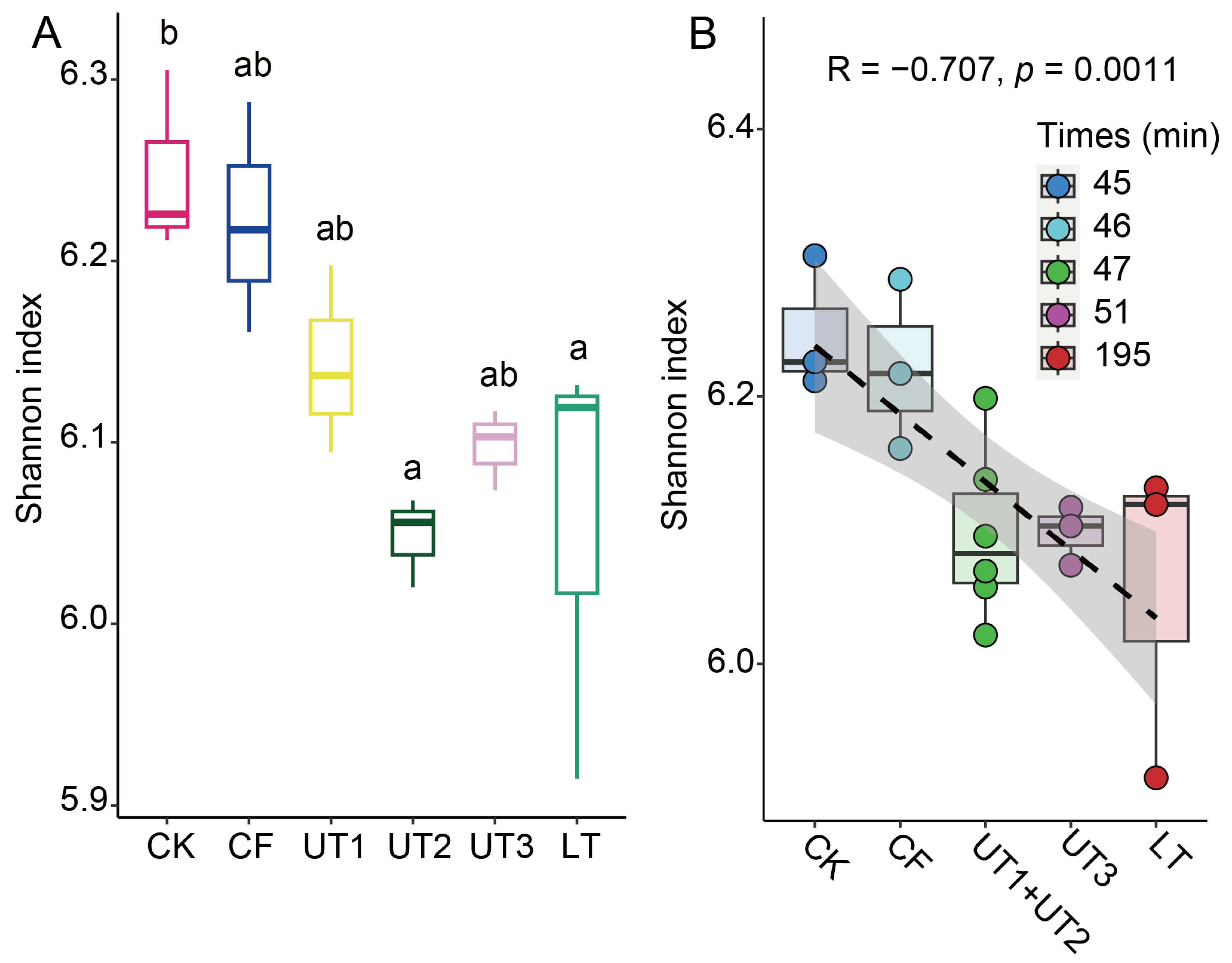
3.2. Ultrasonication Was the Main Factor Impacting the Composition and Diversity of the Extracted Microbiota
3.3. Sonication Time and Strategy Impact Culturable Bacteria Diversity
3.4. The Sonication Strategy Impacts Microbiota Composition during Extraction
3.5. The Processing Time Significantly Decreased Extracted Microbiota α-Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Natarajan, A.; Bhatt, A.S. Microbes and microbiomes in 2020 and beyond. Nat. Commun. 2020, 11, 4988. [Google Scholar] [CrossRef]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Clevers, H. Organoids and organs-on-chips: Insights into human gut-microbe interactions. Cell Host Microbe 2021, 29, 867–878. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lee, Y.; Yu, H.E.; Cho, I.J.; Kang, M.; Lee, S. Sustainable production and degradation of plastics using microbes. Nat. Microbiol. 2023, 8, 2253–2276. [Google Scholar] [CrossRef]
- Jansson, J.K.; McClure, R.; Egbert, R.G. Soil microbiome engineering for sustainability in a changing environment. Nat. Biotechnol. 2023, 41, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Airola, C.; Severino, A.; Porcari, S.; Fusco, W.; Mullish, B.H.; Gasbarrini, A.; Cammarota, G.; Ponziani, F.R.; Ianiro, G. Future Modulation of Gut Microbiota: From Eubiotics to FMT, Engineered Bacteria, and Phage Therapy. Antibiotics 2023, 12, 868. [Google Scholar] [CrossRef]
- Akbar, S.; Gu, L.; Sun, Y.F.; Zhang, L.; Lyu, K.; Huang, Y.; Yang, Z. Understanding host-microbiome-environment interactions: Insights from Daphnia as a model organism. Sci. Total Environ. 2022, 808, 152093. [Google Scholar] [CrossRef]
- Das, S.; Bernasconi, E.; Koutsokera, A.; Wurlod, D.A.; Tripathi, V.; Bonilla-Rosso, G.; Aubert, J.; Derkenne, M.F.; Mercier, L.; Pattaroni, C.; et al. A prevalent and culturable microbiota links ecological balance to clinical stability of the human lung after transplantation. Nat. Commun. 2021, 12, 2126. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef]
- Nirmalkar, K.; Qureshi, F.; Kang, D.K.; Hahn, J.; Adams, J.B.; Rosa, K.B. Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy. Int. J. Mol. Sci. 2022, 23, 13481. [Google Scholar] [CrossRef]
- Yang, Q.; Wei, Y.M.; Zhu, Y.H.; Guo, J.J.; Zhang, J.Y.; He, Y.J.; Li, X.; Liu, J.; Zhou, W. The Interaction between Gut Microbiota and Host Amino Acids Metabolism in Multiple Myeloma. Cancers 2023, 15, 1942. [Google Scholar] [CrossRef]
- Hsu, M.; Tun, K.M.; Batra, K.; Haque, L.; Vongsavath, T.; Hong, A.S. Safety and Efficacy of Fecal Microbiota Transplantation in Treatment of Inflammatory Bowel Disease in the Pediatric Population: A Systematic Review and Meta-Analysis. Microorganisms 2023, 11, 1272. [Google Scholar] [CrossRef]
- Wirth, U.; Jiang, T.; Schardey, J.; Kratz, K.; Li, M.M.; Schirren, M.; Kühn, F.; Bazhin, A.; Werner, J.; Guba, M.; et al. The Role of Microbiota in Liver Transplantation and Liver Transplantation-Related Biliary Complications. Int. J. Mol. Sci. 2023, 24, 4841. [Google Scholar] [CrossRef] [PubMed]
- Bou Zerdan, M.; Niforatos, S.; Nasr, S.; Nasr, D.; Ombada, M.; John, S.; Dutta, D.; Lim, S.H. Fecal microbiota transplant for hematologic and oncologic diseases: Principle and practice. Cancers 2022, 14, 691. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Choi, Y.J.; Kim, H.B.; Chung, H.J.; Hong, S.Y. Identification of the Intestinal Microbes Associated with Locomotion. Int. J. Mol. Sci. 2023, 24, 11392. [Google Scholar] [CrossRef]
- Vanessa, S. Liver-Gut-Interaction: Role of Microbiome Transplantation in the Future Treatment of Metabolic Disease. J. Pers. Med. 2023, 13, 220. [Google Scholar]
- Matheson, J.A.; Damian, H.R.M. The Role of Fecal Microbiota Transplantation in the Treatment of Neurodegenerative Diseases: A Review. Int. J. Mol. Sci. 2023, 24, 1001. [Google Scholar] [CrossRef]
- Nadarajah, K.; Abdul Rahman, N.S.N. The Microbial Connection to Sustainable Agriculture. Plants 2023, 12, 2307. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zhou, H.B.; Yang, Z.Y.; Wang, X.; Chen, H.N.; Zhong, L.; Zheng, W.T.; Niu, W.J.; Wang, S.; Ren, X.M.; et al. Rational construction of genome-reduced Burkholderiales chassis facilitates efficient heterologous production of natural products from proteobacteria. Nat. Commun. 2021, 12, 4347. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, Y.; Gan, G.; Li, W.; Wan, W.; Jiang, Y.; Yang, T.J.; Zhang, Y.; Xu, Y.C.; Wang, Y.K.; et al. Exploring rhizo-microbiome transplants as a tool for protective plant-microbiome manipulation. ISME Commun. 2022, 2, e00094-8. [Google Scholar] [CrossRef] [PubMed]
- Enagbonma, B.J.; Fadiji, A.E.; Ayangbenro, A.S.; Babalola, O.O. Communication between Plants and Rhizosphere Microbiome: Exploring the Root Microbiome for Sustainable Agriculture. Microorganisms 2023, 11, 2003. [Google Scholar] [CrossRef]
- Anguita-Maeso, M.; Navas-Cortés, J.A.; Landa, B.B. Insights into the Methodological, Biotic and Abiotic Factors Influencing the Characterization of Xylem-Inhabiting Microbial Communities of Olive Trees. Plants 2023, 12, 912. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.H.; Wang, M.Y.; Sui, X.; Frey, B.; Liu, Y.N.; Zhang, R.T.; Ni, H.W.; Li, M.H. High Ammonium Addition Changes the Diversity and Structure of Bacterial Communities in Temperate Wetland Soils of Northeastern China. Microorganisms 2023, 11, 2033. [Google Scholar] [CrossRef] [PubMed]
- Abakumov, E.; Zverev, A.; Andronov, E.; Nizamutdinov, T.N. Microbial Composition of Natural, Agricultural, and Technogenic Soils of Both Forest and Forest-Tundra of the Russian North. Appl. Sci. 2023, 13, 8981. [Google Scholar] [CrossRef]
- Chen, Y.L.; Yao, Z.M.; Sun, Y.; Wang, E.Z.; Tian, C.J.; Sun, Y.; Liu, J.; Sun, C.Y.; Tian, L. Current Studies of the Effects of Drought Stress on Root Exudates and Rhizosphere Microbiomes of Crop Plant Species. Int. J. Mol. Sci. 2022, 23, 2374. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.X.; Wu, C.F.; Wang, F.; Lyu, H.; Lu, Y.W.; Yan, C.Q.; Chen, J.P.; Deng, Y.W.; Ge, T.D. Microbial community changes in different underground compartments of potato affected yield and quality. 3 Biotech 2022, 12, 106. [Google Scholar] [CrossRef]
- Maron, P.A.; Sarr, A.; Kaisermann, A.; Lévêque, J.; Mathieu, O.; Guigue, J.; Karimi, B.; Bernard, L.; Dequiedt, S.; Terrat, S.; et al. High microbial diversity promotes soil ecosystem functioning. Appl. Environ. Microbiol. 2018, 84, e02738-17. [Google Scholar] [CrossRef] [PubMed]
- Li, P.D.; Zhu, Z.R.; Zhang, Y.; Xu, J.; Wang, H.; Wang, Z.; Li, H. The phyllosphere microbiome shifts toward combating melanose pathogen. Microbiome 2022, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Wicaksono, W.A.; Li, C.; Michl, K.; Berg, G.; Wang, D.; Xiao, Y.L.; Huang, R.Y.; Kang, H.X.; Zhang, D.Y.; et al. Recovery of metagenome-assembled genomes from the phyllosphere of 110 rice genotypes. Sci. Data 2022, 9, 254. [Google Scholar] [CrossRef]
- Watanabe, M.; Igarashi, K.; Kato, S.; Kamagata, Y.; Kitagawaa, W. Critical Effect of H2O2 in the Agar Plate on the Growth of Laboratory and Environmental Strains. Microbiol. Spectr. 2022, 10, e03336-22. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Caporaso, J.G. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Oksanen, J. Vegan: Community Ecology, Package-R Version 2011.1.17-8. Available online: http://CRAN.R-project.org/package=vegan (accessed on 1 January 2024).
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Deng, S.; Wipf, H.M.L.; Pierroz, G.; Raab, T.K.; Khanna, R.; Coleman-Derr, D. A plant growth-promoting microbial soil amendment dynamically alters the strawberry root bacterial microbiome. Sci. Rep. 2019, 9, 17677. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.; DeGraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 2017, 11, 2691–2704. [Google Scholar] [CrossRef]
- Sharma, P.; Thakur, D. Antimicrobial biosynthetic potential and diversity of culturable soil actinobacteria from forest ecosystems of Northeast India. Sci. Rep. 2020, 10, 4104. [Google Scholar] [CrossRef] [PubMed]
- Campolino, M.L.; de Paula Lana, U.G.; Gomes, E.A.; Coelho, A.M.; de Sousa, S.M. Phosphate fertilization affects rhizosphere microbiome of maize and sorghum genotypes. Braz. J. Microbiol. 2022, 53, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Ismaïl, R.; Aviat, F.; Michel, V.; Le Bayon, I.; Gay-Perret, P.; Kutnik, M.; Fédérighi, M. Methods for recovering microorganisms from solid surfaces used in the food industry: A review of the literature. Int. J. Environ. Res. Public Health 2013, 10, 6169–6183. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E.C. The Function of Sonication in the Diagnosis of Periprosthetic Joint Infection After Total Knee Arthroplasty. Arch. Bone Jt. Surg. 2022, 10, 735. [Google Scholar]
- Broekman, S.; Pohlmann, O.; Beardwood, E.S.; de Meulenaer, E.C. Ultrasonic treatment for microbiological control of water systems. Ultrason. Sonochemistry 2010, 17, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, S.; Cao, P. Synergistic bactericidal effects and mechanisms of low intensity ultrasound and antibiotics against bacteria: A review. Ultrason. Sonochemistry 2012, 19, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Lara-Gutiérrez, J.; Stocker, R. Environmental fluctuations and their effects on microbial communities, populations and individuals. FEMS Microbiol. Rev. 2021, 45, fuaa068. [Google Scholar] [CrossRef] [PubMed]
- Chepsergon, J.; Moleleki, L.N. Rhizosphere bacterial interactions and impact on plant health. Curr. Opin. Microbiol. 2023, 73, 102297. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Read, J.S.; Welkie, D.G.; Kratz, T.K.; Wu, C.H.; McMahon, K.D. Resistance, resilience and recovery: Aquatic bacterial dynamics after water column disturbance. Environ. Microbiol. 2011, 13, 2752–2767. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; McMahon, K.D. The influence of habitat heterogeneity on freshwater bacterial community composition and dynamics. Environ. Microbiol. 2008, 10, 1057–1067. [Google Scholar] [CrossRef]
- BIG Data Center Members. Database resources of the BIG data center in 2018. Nucleic Acids Res. 2018, 46, D14–D20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method Name | Oscillation (200 rpm) | Sonication (30 kHz) | Centrifugation (600 rpm, 4 °C) |
|---|---|---|---|
| CK | 30 min | - | - |
| CF | 30 min | - | 1 min |
| UT1 | 30 min | 2 min | - |
| LT | 180 min | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhang, C.; Ma, J.; Peng, Q.; Du, X.; Sun, S.; Cheng, J.; Peng, W.; Chen, L.; Gu, Z.; et al. Extraction Methods Determine the Quality of Soil Microbiota Acquisition. Microorganisms 2024, 12, 403. https://doi.org/10.3390/microorganisms12020403
Liu Z, Zhang C, Ma J, Peng Q, Du X, Sun S, Cheng J, Peng W, Chen L, Gu Z, et al. Extraction Methods Determine the Quality of Soil Microbiota Acquisition. Microorganisms. 2024; 12(2):403. https://doi.org/10.3390/microorganisms12020403
Chicago/Turabian StyleLiu, Zhuoxin, Chi Zhang, Jiejia Ma, Qianze Peng, Xiaohua Du, Shu’e Sun, Ju’e Cheng, Weiye Peng, Lijie Chen, Zepei Gu, and et al. 2024. "Extraction Methods Determine the Quality of Soil Microbiota Acquisition" Microorganisms 12, no. 2: 403. https://doi.org/10.3390/microorganisms12020403