
Deciphering Microbial Communities and Distinct Metabolic Pathways in the Tangyin Hydrothermal Fields of Okinawa Trough through Metagenomic and Genomic Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
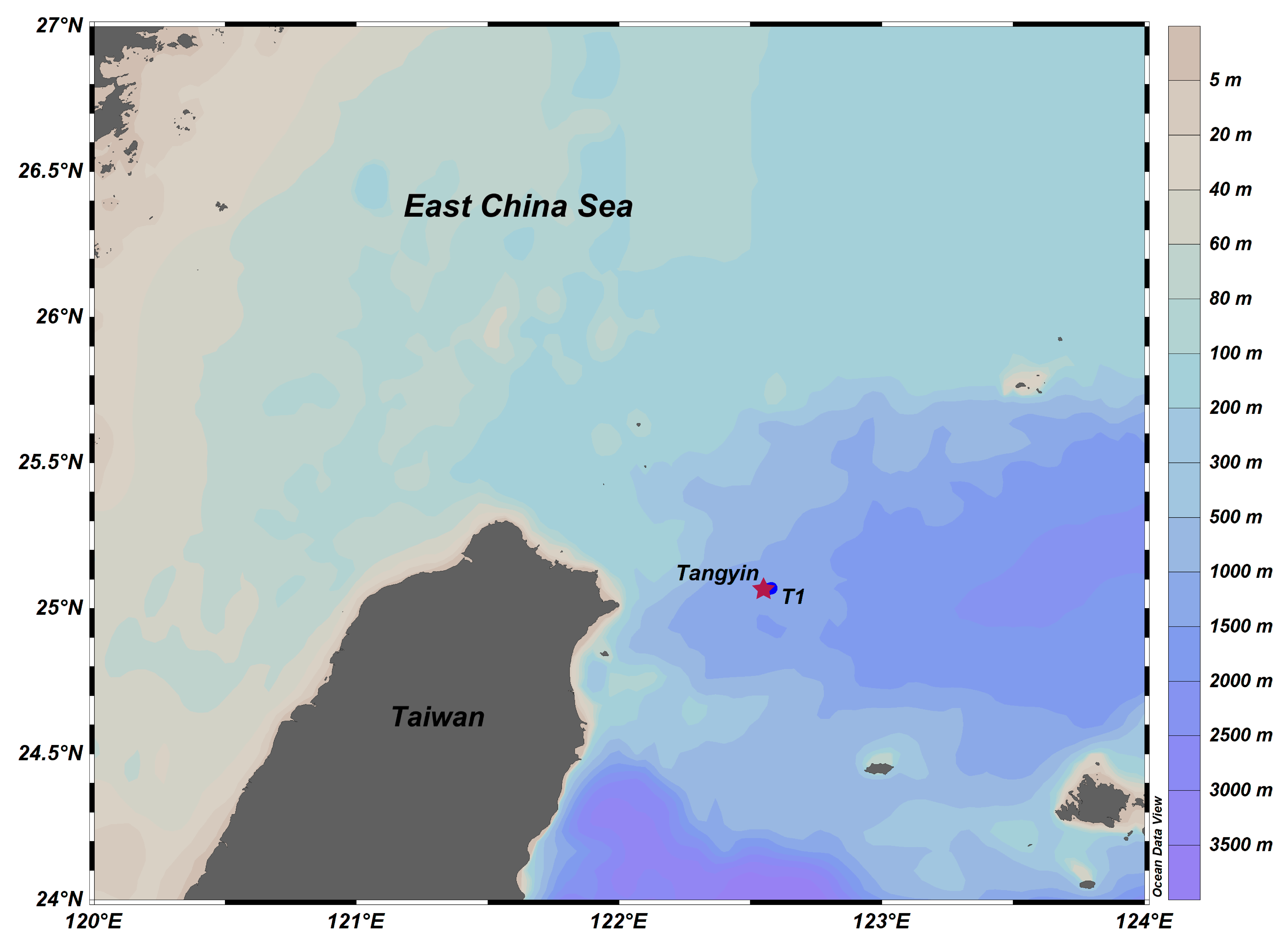
2.1. Sampling Site and DNA and RNA Extraction
2.2. DNA and RNA Based on 16S rRNA Gene Amplicon Sequencing
2.3. Gene Functional Classification of the Metagenome
2.4. Metagenomic Binning, Taxonomic Assignment, and Functional Characterization
3. Results
3.1. Taxonomic Compositions by the 16S rRNA Gene and Metagenomic Gene
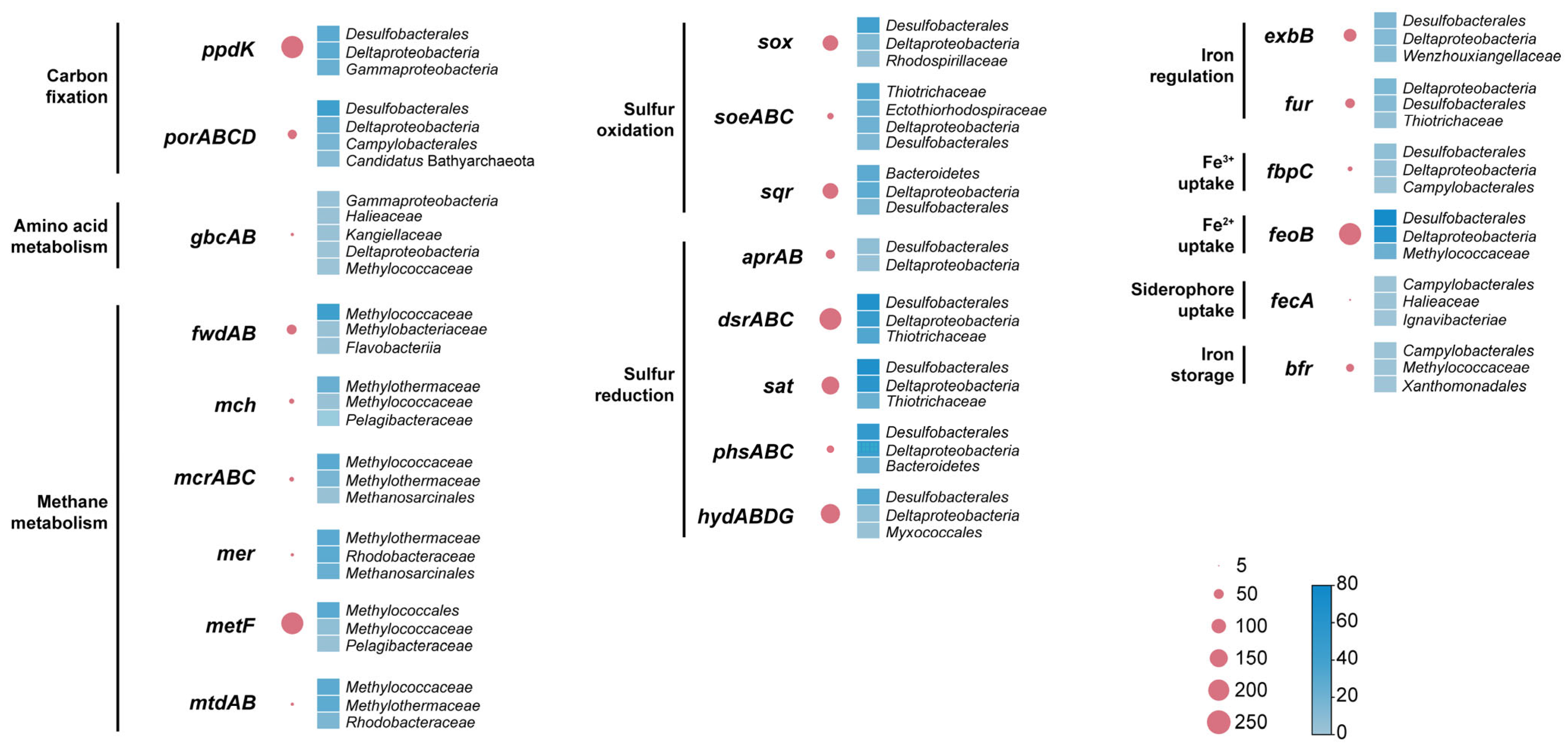
3.2. Genes Involved in Carbon, Sulfur, and Iron Metabolisms
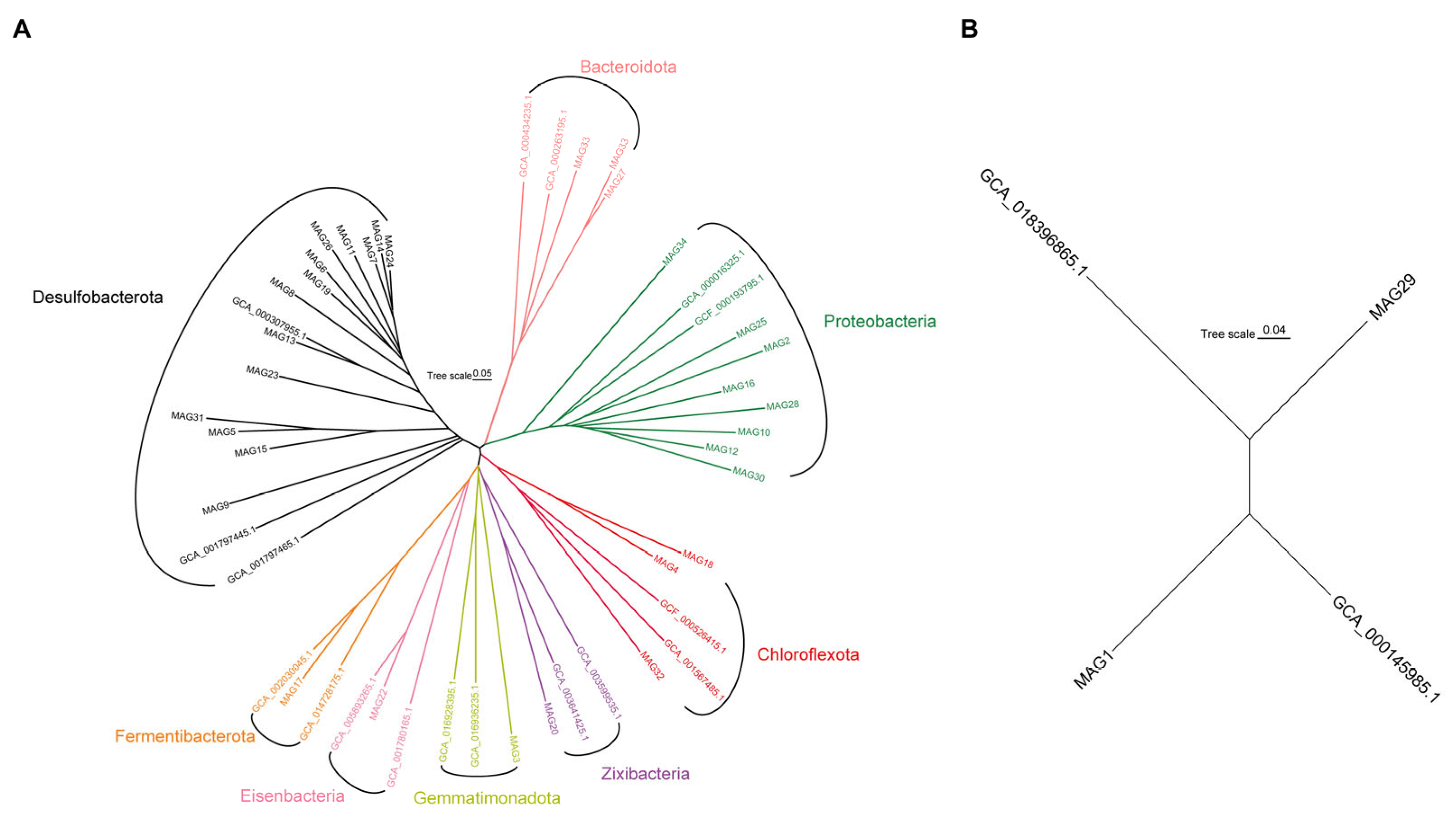
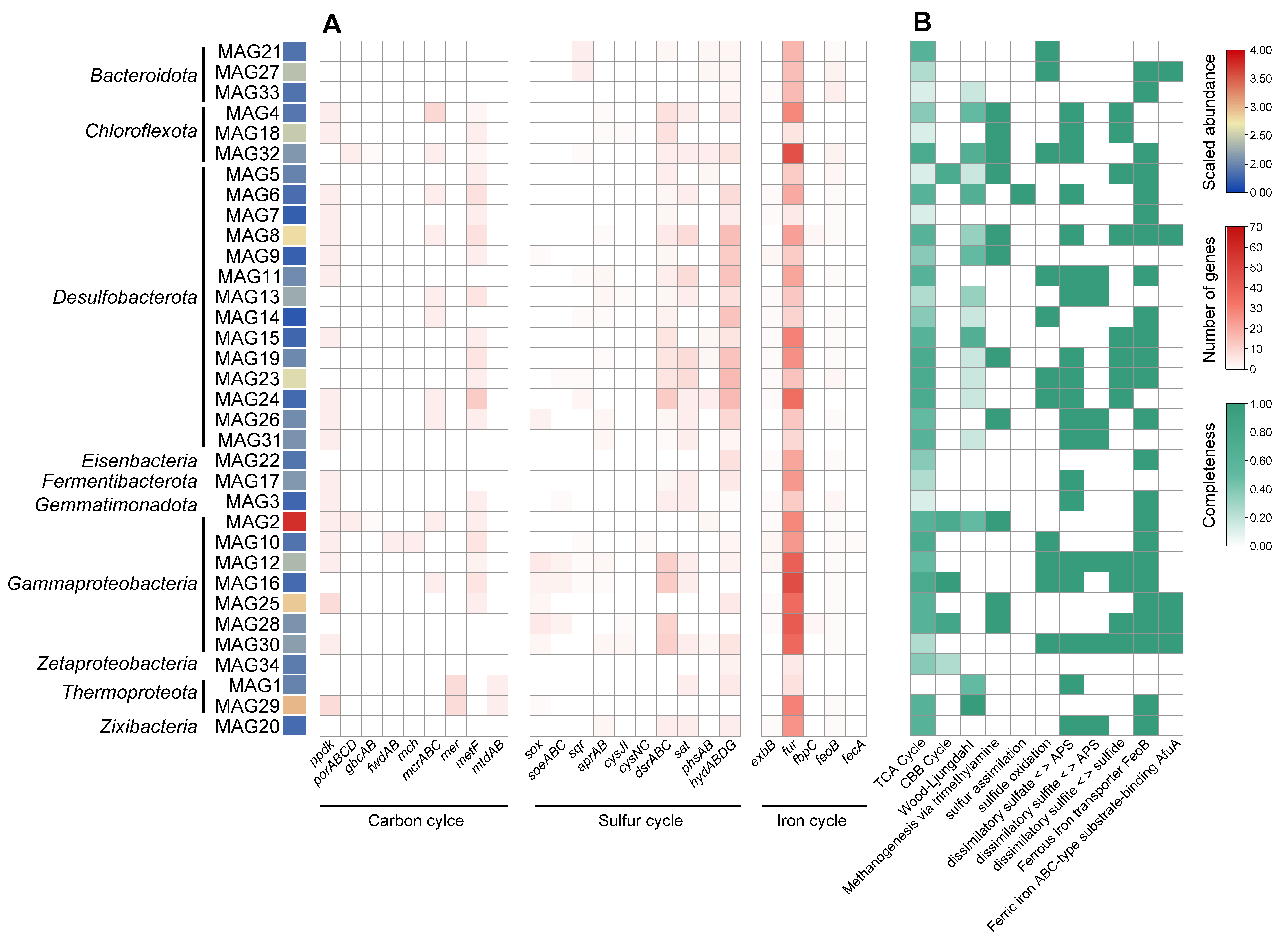
3.3. Taxonomic Assignments and Carbon, Sulfur, and Iron Metabolisms of MAGs
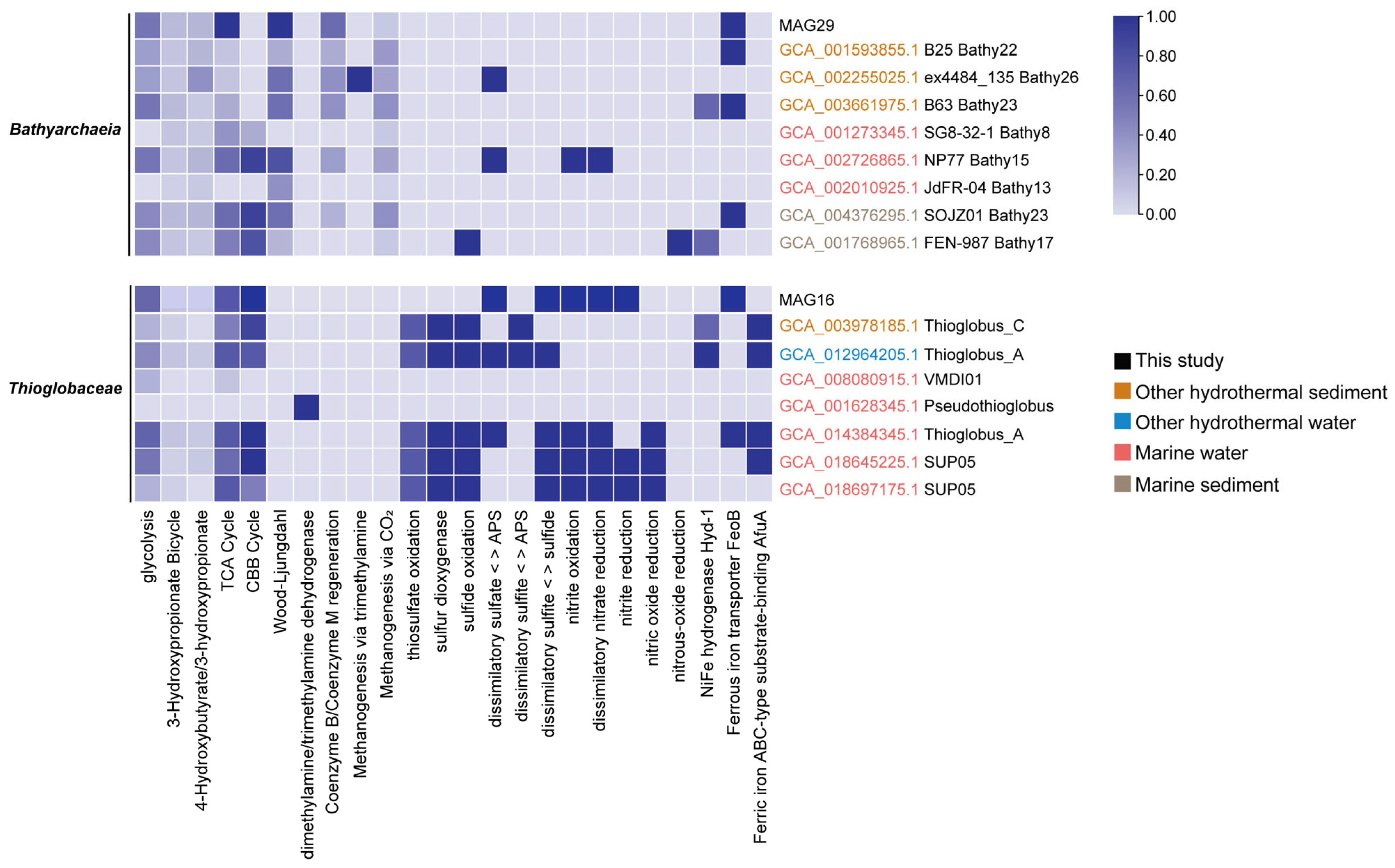
3.4. The Unique Metabolic Features of Representative MAGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dick, G.J. The microbiomes of deep-sea hydrothermal vents: Distributed globally, shaped locally. Nat. Rev. Microbiol. 2019, 17, 271–283. [Google Scholar] [CrossRef]
- Lonsdale, P. Clustering of suspension-feeding macrobenthos near abyssal hydrothermal vents at oceanic spreading centers. Deep Sea Res. 1977, 24, 857–863. [Google Scholar] [CrossRef]
- Miyazaki, J.; Kawagucci, S.; Makabe, A.; Takahashi, A.; Kitada, K.; Torimoto, J.; Matsui, Y.; Tasumi, E.; Shibuya, T.; Nakamura, K.; et al. Deepest and hottest hydrothermal activity in the Okinawa Trough: The Yokosuka site at Yaeyama Knoll. R. Soc. Open Sci. 2017, 4, 171570. [Google Scholar] [CrossRef] [PubMed]
- Polinski, J.M.; Rodrigue, M.; Meyer, J.D.; Harke, M.J. Drifting in the deep: Metatranscriptomics and metabarcoding reveal sustained metabolic activity and community composition in hydrothermal vent plume microbial communities. Front. Mar. Sci. 2023, 10, 1219784. [Google Scholar] [CrossRef]
- Mordukhovich, V.V.; Krylova, E.M.; Dando, P.R. Seeps and vents of the Bering Sea. Deep-Sea Res. Part II-Top. Stud. Oceanogr. 2023, 209, 105290. [Google Scholar] [CrossRef]
- Janssen, D.J.; Gilliard, D.; Rickli, J.; Nasemann, P.; Koschinsky, A.; Hassler, C.S.; Bowie, A.R.; Ellwood, M.J.; Kleint, C.; Jaccard, S.L. Chromium stable isotope distributions in the southwest Pacific Ocean and constraints on hydrothermal input from the Kermadec Arc. Geochim. Cosmochim. Acta 2023, 342, 31–44. [Google Scholar] [CrossRef]
- Zeng, Z.; Chen, S.; Ma, Y.; Yin, X.; Wang, X.; Zhang, S.; Zhang, J.; Wu, X.; Li, Y.; Dong, D.; et al. Chemical compositions of mussels and clams from the Tangyin and Yonaguni Knoll IV hydrothermal fields in the southwestern Okinawa Trough. Ore Geol. Rev. 2017, 87, 172–191. [Google Scholar] [CrossRef]
- Meier, D.V.; Pjevac, P.; Bach, W.; Hourdez, S.; Girguis Peter, R.; Vidoudez, C.; Amann, R.; Meyerdierks, A. Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 2017, 11, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, Y.; Lin, H.; Wang, X.; Li, M.; Liu, Y.; Yu, M.; Zhao, M.; Pedentchouk, N.; Lea-Smith, D.J. Proliferation of hydrocarbon-degrading microbes at the bottom of the Mariana Trench. Microbiome 2019, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, C.; Kirkpatrick, J.B.; D’Hondt, S.; Loose, B. Methane-oxidizing seawater microbial communities from an Arctic shelf. Biogeosciences 2018, 15, 3311–3329. [Google Scholar] [CrossRef]
- Nunoura, T.; Takaki, Y.; Kazama, H.; Kakuta, J.; Shimamura, S.; Makita, H.; Hirai, M.; Miyazaki, M.; Takai, K. Physiological and genomic features of a novel sulfur-oxidizing gammaproteobacterium belonging to a previously uncultivated symbiotic lineage isolated from a hydrothermal vent. PLoS ONE 2014, 9, e104959. [Google Scholar] [CrossRef]
- Amrani, A.; Bergon, A.; Holota, H.; Tamburini, C.; Garel, M.; Ollivier, B.; Imbert, J.; Dolla, A.; Pradel, N. Transcriptomics reveal several gene expression patterns in the piezophile Desulfovibrio hydrothermalis in response to hydrostatic pressure. PLoS ONE 2014, 9, e106831. [Google Scholar] [CrossRef]
- Cao, H.; Wang, Y.; Lee, O.O.; Zeng, X.; Shao, Z.; Qian, P.Y. Microbial sulfur cycle in two hydrothermal chimneys on the Southwest Indian Ridge. mBio 2014, 5, e00980-13. [Google Scholar] [CrossRef]
- Tao, C.; Guo, Z.; Liang, J.; Ding, T.; Yang, W.; Liao, S.; Chen, M.; Zhou, F.; Chen, J.; Wang, N. Sulfide metallogenic model for the ultraslow-spreading Southwest Indian Ridge. Sci. China Earth Sci. 2023, 66, 1212–1230. [Google Scholar] [CrossRef]
- Ishibashi, J.-I.; Ikegami, F.; Tsuji, T.; Urabe, T. Hydrothermal activity in the Okinawa Trough back-arc basin: Geological background and hydrothermal mineralization. In Subseafloor Biosphere Linked to Hydrothermal Systems; Springer: Berlin/Heidelberg, Germany, 2015; pp. 337–359. [Google Scholar] [CrossRef]
- Takai, K.; Mottl, M.J.; Nielsen, S.H. IODP Expedition 331: Strong and expansive subseafloor hydrothermal activities in the Okinawa Trough. Sci. Drill. 2012, 13, 19–27. [Google Scholar] [CrossRef]
- Toki, T.; Itoh, M.; Iwata, D.; Ohshima, S.; Shinjo, R.; Ishibashi, J.-I.; Tsunogai, U.; Takahata, N.; Sano, Y.; Yamanaka, T. Geochemical characteristics of hydrothermal fluids at Hatoma Knoll in the southern Okinawa Trough. Geochem. J. 2016, 50, 493–525. [Google Scholar] [CrossRef]
- Wang, L.; Yu, M.; Liu, Y.; Liu, J.; Wu, Y.; Li, L.; Liu, J.; Wang, M.; Zhang, X.-H. Comparative analyses of the bacterial community of hydrothermal deposits and seafloor sediments across Okinawa Trough. J. Mar. Syst. 2018, 180, 162–172. [Google Scholar] [CrossRef]
- Zhang, X.; Zhai, S.; Yu, Z.; Wang, S.; Cai, Z. Mineralogy and geological significance of hydrothermal deposits from the Okinawa trough. J. Mar. Syst. 2016, 180, 124–131. [Google Scholar] [CrossRef]
- Yanagawa, K.; Breuker, A.; Schippers, A.; Nishizawa, M.; Ijiri, A.; Hirai, M.; Takaki, Y.; Sunamura, M.; Urabe, T.; Nunoura, T.; et al. Microbial community stratification controlled by the subseafloor fluid flow and geothermal gradient at the Iheya North hydrothermal field in the Mid-Okinawa Trough (Integrated Ocean Drilling Program Expedition 331). Appl. Environ. Microbiol. 2014, 80, 6126–6135. [Google Scholar] [CrossRef] [PubMed]
- Nunoura, T.; Oida, H.; Nakaseama, M.; Kosaka, A.; Ohkubo, S.B.; Kikuchi, T.; Kazama, H.; Hosoi-Tanabe, S.; Nakamura, K.; Kinoshita, M.; et al. Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the Yonaguni Knoll IV hydrothermal field in the Southern Okinawa Trough. Appl. Environ. Microbiol. 2010, 76, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Inagaki, F. A comparative study of microbial diversity and community structure in marine sediments using poly(A) tailing and reverse transcription-PCR. Front. Microbiol. 2013, 4, 160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Q.L.; Zeng, Z.G.; Chen, S.; Sun, L. Microbial diversity in the deep-sea sediments of Iheya North and Iheya Ridge, Okinawa Trough. Microbiol. Res. 2015, 177, 43–52. [Google Scholar] [CrossRef]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Shi, X.; Cheng, H.; Yao, P.; Zhao, B.; Chen, L.; Liu, J.; Cao, L.; Wang, M.; Fu, L.; et al. Horizontal and vertical heterogeneity of sediment microbial community in Site F cold seep, the South China Sea. Front. Mar. Sci. 2022, 9, 957762. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.; Land, M.; Larimer, F.; Hauser, L. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Li, W.; Adam, G. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Xue, C.-X.; Liu, J.; Lea-Smith, D.J.; Rowley, G.; Lin, H.; Zheng, Y.; Zhu, X.-Y.; Liang, J.; Ahmad, W.; Todd, J.D.; et al. Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms 2020, 8, 1309. [Google Scholar] [CrossRef]
- Peng, X.; Wang, J.; Zhang, Z.; Xiao, Q.; Li, M. Re-alignment of the unmapped reads with base quality score. BMC Bioinform. 2015, 16, S8. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab: Berkeley, CA, USA, 2014. [Google Scholar]
- Gautam, A.; Felderhoff, H.; Bağci, C.; Huson, D.H. Using AnnoTree to get more assignments, faster, in DIAMOND+MEGAN microbiome analysis. bioRxiv 2021, 7, e01408-21. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; Di Ruggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- David, M.E.; Steven, K. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, 1319. [Google Scholar] [CrossRef]
- Graham, E.D.; Heidelberg, J.F.; Tully, B.J. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 2018, 12, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Teske, A. The Guaymas Basin—A hot spot for hydrothermal generation and anaerobic microbial degradation of hydrocarbons. Int. Biodeterior. Biodegrad. 2024, 186, 105700. [Google Scholar] [CrossRef]
- Hedderich, R.; Berkessel, A.; Thauer, R.K. Purification and properties of heterodisulfide reductase from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 1990, 193, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Yu, X.; Zhou, J.; Gu, H.; Ding, J.; Peng, Y.; He, Q.; Tian, Y.; Liu, J.; Wang, S.; et al. MCycDB: A curated database for comprehensively profiling methane cycling processes of environmental microbiomes. Mol. Ecol. Resour. 2022, 22, 1803–1823. [Google Scholar] [CrossRef] [PubMed]
- Sandy, M.; Butler, A. Microbial iron acquisition: Marine and terrestrial siderophores. Chem. Rev. 2009, 109, 4580–4595. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, B.M.; Barbeau, K.A. Iron transporters in marine prokaryotic genomes and metagenomes. Environ. Microbiol. 2012, 14, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.-L.; Evans, P.; Li, Y.; Rao, Y.; Qu, Y.-N.; Tan, S.; Jiao, J.; Chen, Y.-T.; Hedlund, B.; Shu, W.-S.; et al. Comparative Genomics Reveals Thermal Adaptation and a High Metabolic Diversity in “Candidatus Bathyarchaeia”. mSystems 2021, 6, 252–273. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Sauter, A.; Braun, V. Defined inactive FecA derivatives mutated in functional domains of the outer membrane transport and signaling protein of Escherichia coli K-12. J. Bacteriol. 2004, 186, 5303–5310. [Google Scholar] [CrossRef]
- Andrews, S.C.; Harrison, P.M.; Guest, J.R. Cloning, sequencing, and mapping of the bacterioferritin gene (bfr) of Escherichia coli K-12. J. Bacteriol. 1989, 171, 3940–3947. [Google Scholar] [CrossRef]
- Inagaki, F.; Kuypers, M.M.; Tsunogai, U.; Ishibashi, J.; Nakamura, K.; Treude, T.; Ohkubo, S.; Nakaseama, M.; Gena, K.; Chiba, H.; et al. Microbial community in a sediment-hosted CO2 lake of the southern Okinawa Trough hydrothermal system. Proc. Natl. Acad. Sci. USA 2006, 103, 14164–14169. [Google Scholar] [CrossRef]
- Eick-Helmerich, K.; Hantke, K.; Braun, V. Cloning and expression of the exbB gene of Escherichia coli K-12. Mol. Gen. Genet. 1987, 206, 246–251. [Google Scholar] [CrossRef]
- Nunoura, T.; Takai, K. Comparison of microbial communities associated with phase-separation-induced hydrothermal fluids at the Yonaguni Knoll IV hydrothermal field, the Southern Okinawa Trough. FEMS Microbiol. Ecol. 2009, 67, 351–370. [Google Scholar] [CrossRef]
- Aoyama, S.; Nishizawa, M.; Takai, K.; Ueno, Y. Microbial sulfate reduction within the Iheya North subseafloor hydrothermal system constrained by quadruple sulfur isotopes. Earth Planet. Sci. Lett. 2014, 398, 113–126. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L. Comparative metagenomic analysis of the microbial communities in the surroundings of Iheya north and Iheya ridge hydrothermal fields reveals insights into the survival strategy of microorganisms in deep-sea environments. J. Mar. Syst. 2016, 180, 102–111. [Google Scholar] [CrossRef]
- He, Y.; Xiao, X.; Wang, F. Metagenome reveals potential microbial degradation of hydrocarbon coupled with sulfate reduction in an oil-immersed chimney from Guaymas Basin. Front. Microbiol. 2013, 4, 148. [Google Scholar] [CrossRef] [PubMed]
- Shima, S.; Huang, G.; Wagner, T.; Ermler, U. Structural Basis of Hydrogenotrophic Methanogenesis. Annu. Rev. Microbiol. 2020, 74, 713–733. [Google Scholar] [CrossRef] [PubMed]
- Meister, P.; Herda, G.; Petrishcheva, E.; Gier, S.; Dickens, G.R.; Bauer, C.; Liu, B. Microbial Alkalinity Production and Silicate Alteration in Methane Charged Marine Sediments: Implications for Porewater Chemistry and Diagenetic Carbonate Formation. Front. Earth Sci. 2022, 9, 18. [Google Scholar] [CrossRef]
- Nakagawa, S.; Takaki, Y. Nonpathogenic Epsilonproteobacteria. In Encyclopedia of Life Sciences; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Yang, Y.; Sanford, R.; Yan, J.; Chen, G.; Cápiro Natalie, L.; Li, X. Roles of Organohalide-Respiring Dehalococcoidia in Carbon Cycling. mSystems 2020, 5, e00757-19. [Google Scholar] [CrossRef] [PubMed]
- Shinsuke, K.; Hitoshi, C.; Jun-Ichiro, I.; Toshiro, Y.; Tomohiro, T.; Yasuyuki, M.; Yuichiro, U.; Akiko, M.; Kazuhiro, I.; Naohiro, Y.; et al. Hydrothermal fluid geochemistry at the Iheya North field in the mid-Okinawa Trough: Implication for origin of methane in subseafloor fluid circulation systems. Geochem. J. 2011, 45, 109–124. [Google Scholar] [CrossRef]
- Khider, M.; Brautaset, T.; Irla, M. Methane monooxygenases: Central enzymes in methanotrophy with promising biotechnological applications. World J. Microbiol. Biotechnol. 2021, 37, 72. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, X.; Lu, R.; Zhou, Y.; Zheng, P.; Feng, D.; Wang, Y. Microbial ecology of sulfur cycling near the sulfate-methane transition of deep-sea cold seep sediments. Environ. Microbiol. 2021, 23, 6844–6858. [Google Scholar] [CrossRef] [PubMed]
- Rubin-Blum, M.; Antony, C.P.; Sayavedra, L.; Martínez-Pérez, C.; Birgel, D.; Peckmann, J.; Wu, Y.C.; Cardenas, P.; Macdonald, I.; Marcon, Y. Fueled by methane: Deep-sea sponges from asphalt seeps gain their nutrition from methane-oxidizing symbionts. ISME J. 2019, 13, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Teske, A.P.; MacGregor, B.J.; McKay, L.J.; Mendlovitz, H.; Albert, D.; Ma, Z.; Li, J. Thermal Selection of Microbial Communities and Preservation of Microbial Function in Guaymas Basin Hydrothermal Sediments. Appl. Environ. Microbiol. 2023, 89, e00018-23. [Google Scholar] [CrossRef]
- Costa, K.C.; Wong, P.M.; Wang, T.; Lie, T.J.; Dodsworth, J.A.; Swanson, I.; Burn, J.A.; Hackett, M.; Leigh, J.A. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc. Natl. Acad. Sci. USA 2010, 107, 11050–11055. [Google Scholar] [CrossRef]
- Li, M.; Toner, B.; Baker, B.; Breier, J.; Sheik, C.; Dick, G. Microbial iron uptake as a mechanism for dispersing iron from deep-sea hydrothermal vents. Nat. Commun. 2014, 5, 3192. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, C.; Peng, Y.; Zhang, H.X.; Shi, L.D.; Wei, G.; Hubert, C.R.J.; Wang, Y.; Greening, C. Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments. Nat. Commun. 2022, 13, 4885. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Zhong, C.; Xu, Z.; Lu, G.; Jing, H.; Liu, H. Genomic Insights into Niche Partitioning across Sediment Depth among Anaerobic Methane-Oxidizing Archaea in Global Methane Seeps. mSystems 2023, 8, e01179-22. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Bowie, A.R.; Boyd, P.W.; Buck, K.N.; Johnson, K.S.; Saito, M.A. The integral role of iron in ocean biogeochemistry. Nature 2017, 543, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Chan, C.S.; Barco, R.A.; Merino, N. FeGenie: A Comprehensive Tool for the Identification of Iron Genes and Iron Gene Neighborhoods in Genome and Metagenome Assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Cheng, H.; Yin, F.; Liu, J.; Zhang, X.-H.; Yu, M. Deciphering Microbial Communities and Distinct Metabolic Pathways in the Tangyin Hydrothermal Fields of Okinawa Trough through Metagenomic and Genomic Analyses. Microorganisms 2024, 12, 517. https://doi.org/10.3390/microorganisms12030517
Li J, Cheng H, Yin F, Liu J, Zhang X-H, Yu M. Deciphering Microbial Communities and Distinct Metabolic Pathways in the Tangyin Hydrothermal Fields of Okinawa Trough through Metagenomic and Genomic Analyses. Microorganisms. 2024; 12(3):517. https://doi.org/10.3390/microorganisms12030517
Chicago/Turabian StyleLi, Jiake, Haojin Cheng, Fu Yin, Jiwen Liu, Xiao-Hua Zhang, and Min Yu. 2024. "Deciphering Microbial Communities and Distinct Metabolic Pathways in the Tangyin Hydrothermal Fields of Okinawa Trough through Metagenomic and Genomic Analyses" Microorganisms 12, no. 3: 517. https://doi.org/10.3390/microorganisms12030517