
Evaluation of a Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)-Based Microneutralization Assay for Assessing Clinical Human Cytomegalovirus-Neutralizing Antibody Activity

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Media
2.2. Viruses and Antibodies
2.3. Clinical Specimens
2.4. RT-qPCR Assay
2.5. Immunostaining Assay
2.6. Neutralization Assay
2.7. Comparison of hCMV RNA Preparation with and without RNA Extraction
2.8. Timepoint Optimization for RT-qPCR Analysis
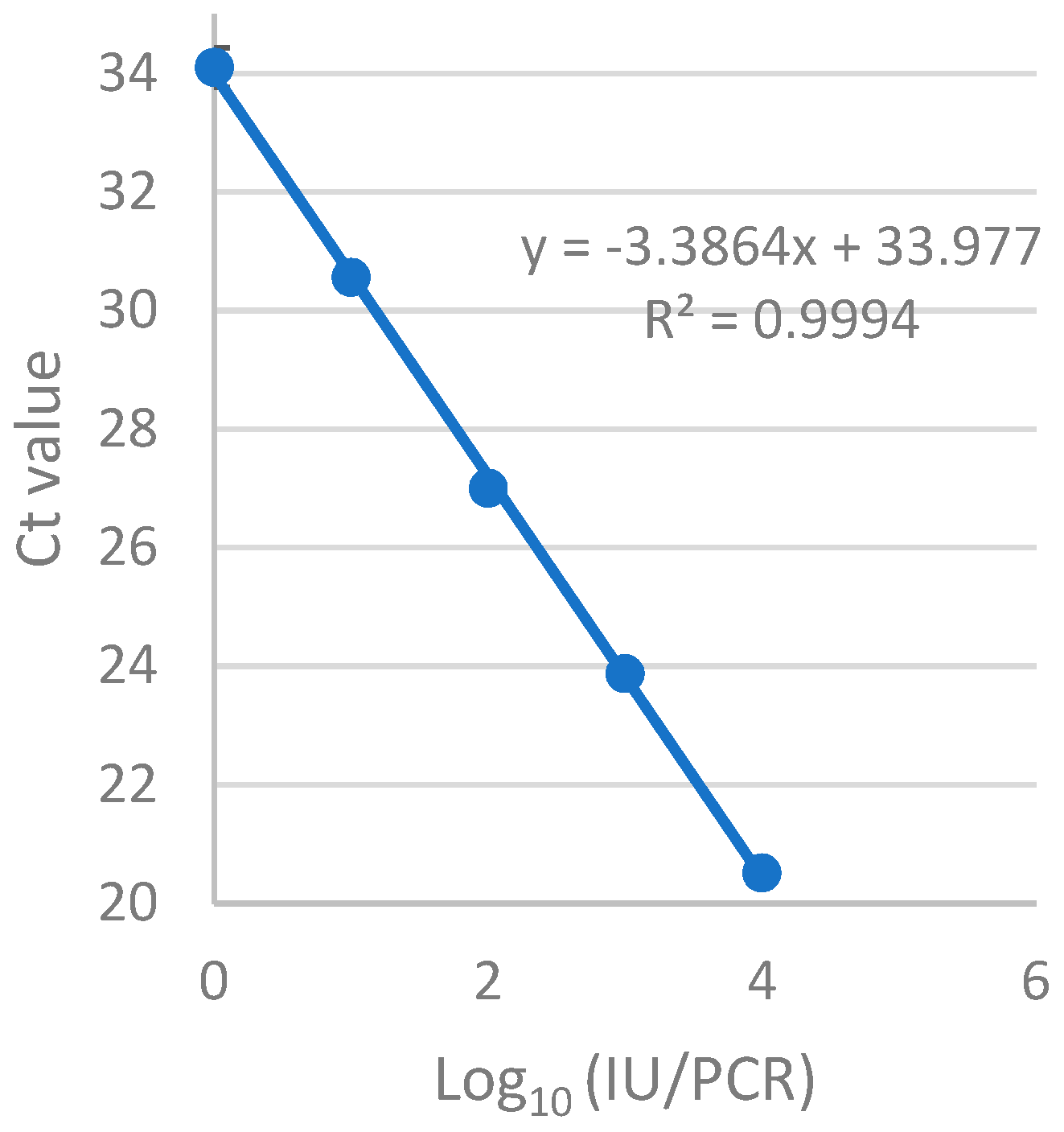
2.9. Evaluation of Sensitivity and Precision of the RT-qPCR Assay
2.10. Comparison between the RT-qPCR and Immunostaining Assays for hCMV Infectivity Detection
2.11. Statistical Analysis
3. Results
3.1. Optimization of the Direct Cell Lysate for RT-qPCR Reaction
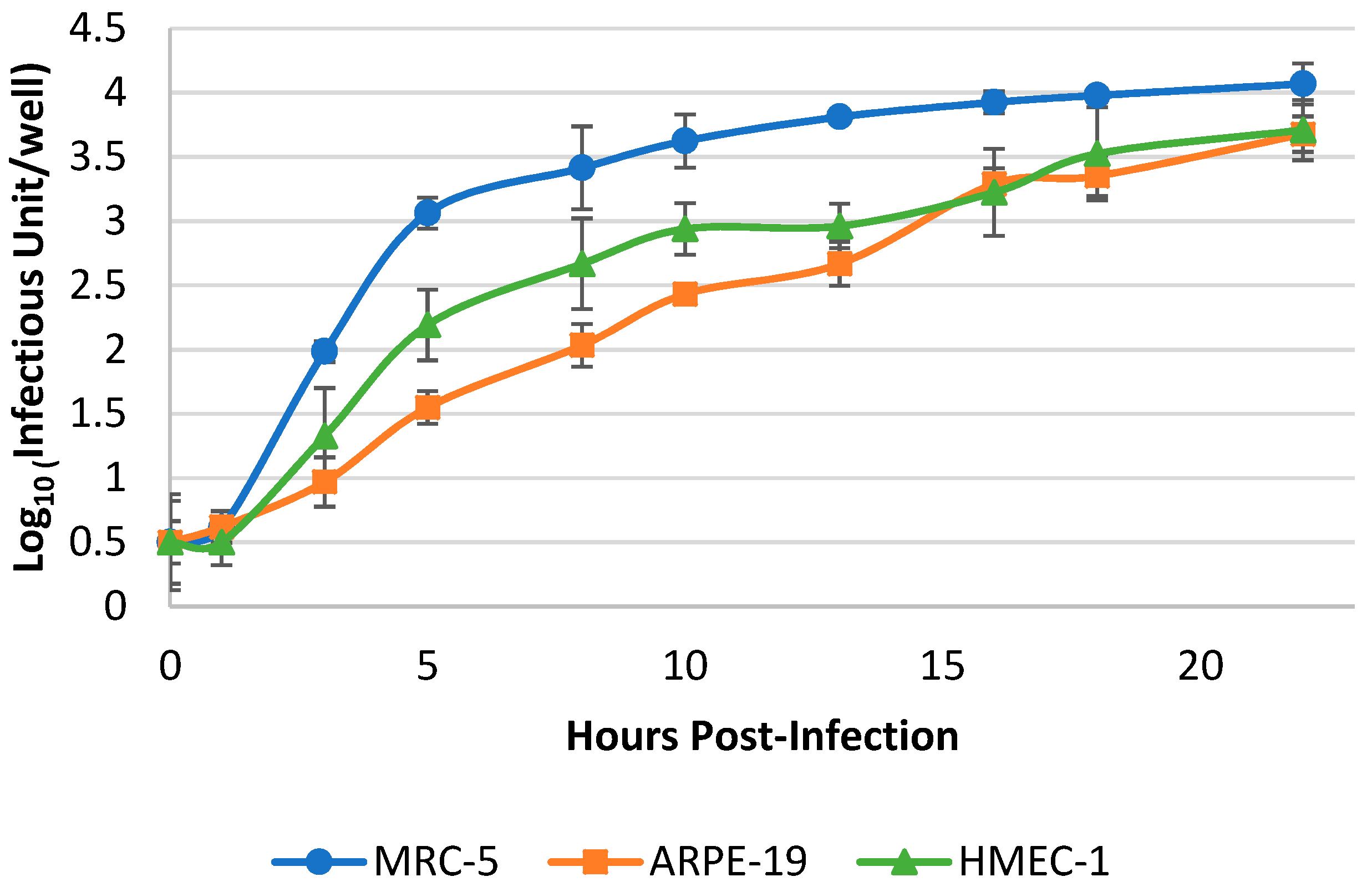
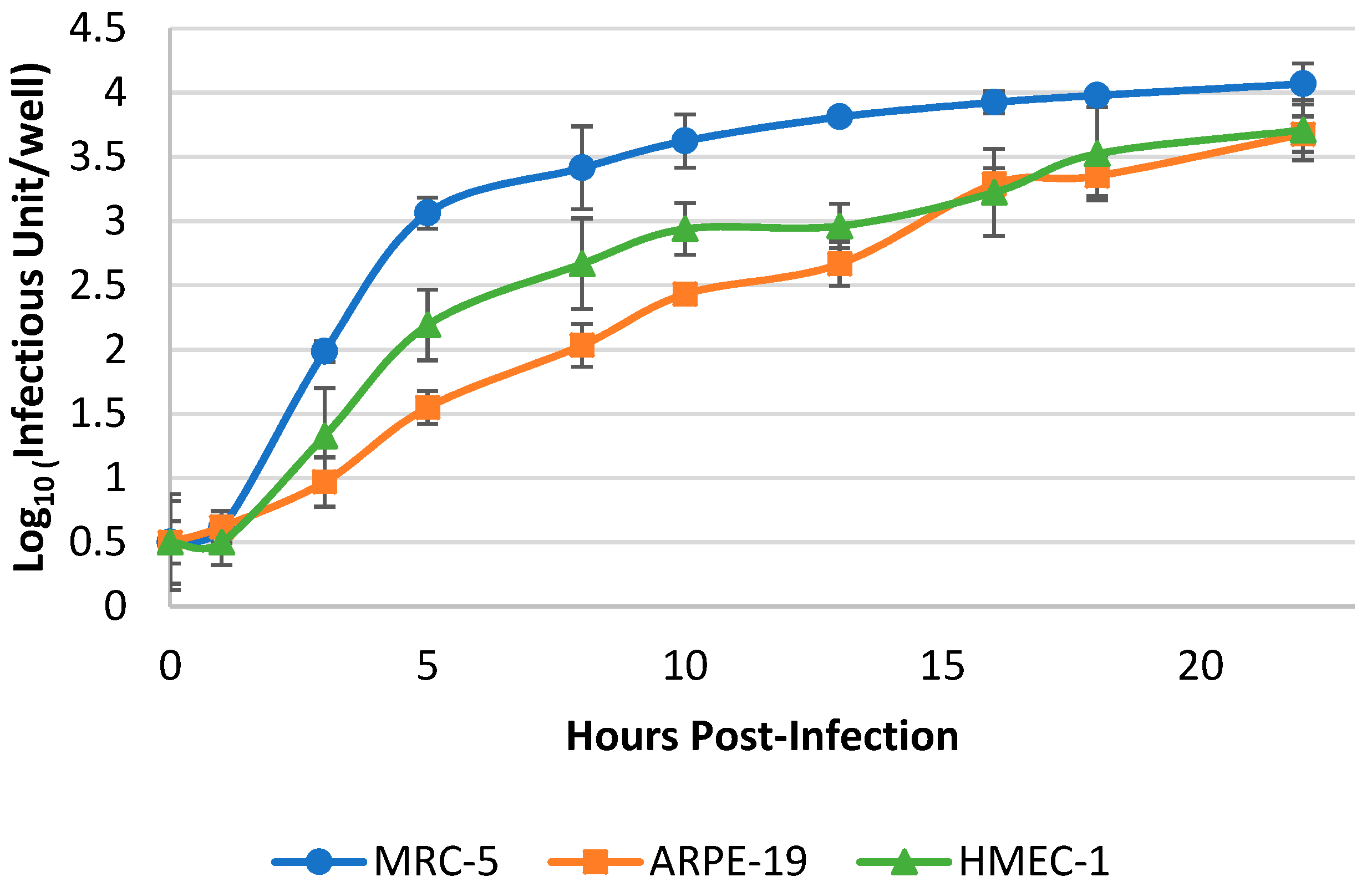
3.2. Optimization of Harvest Timepoint of Cultured hCMV for the RT-qPCR Assay
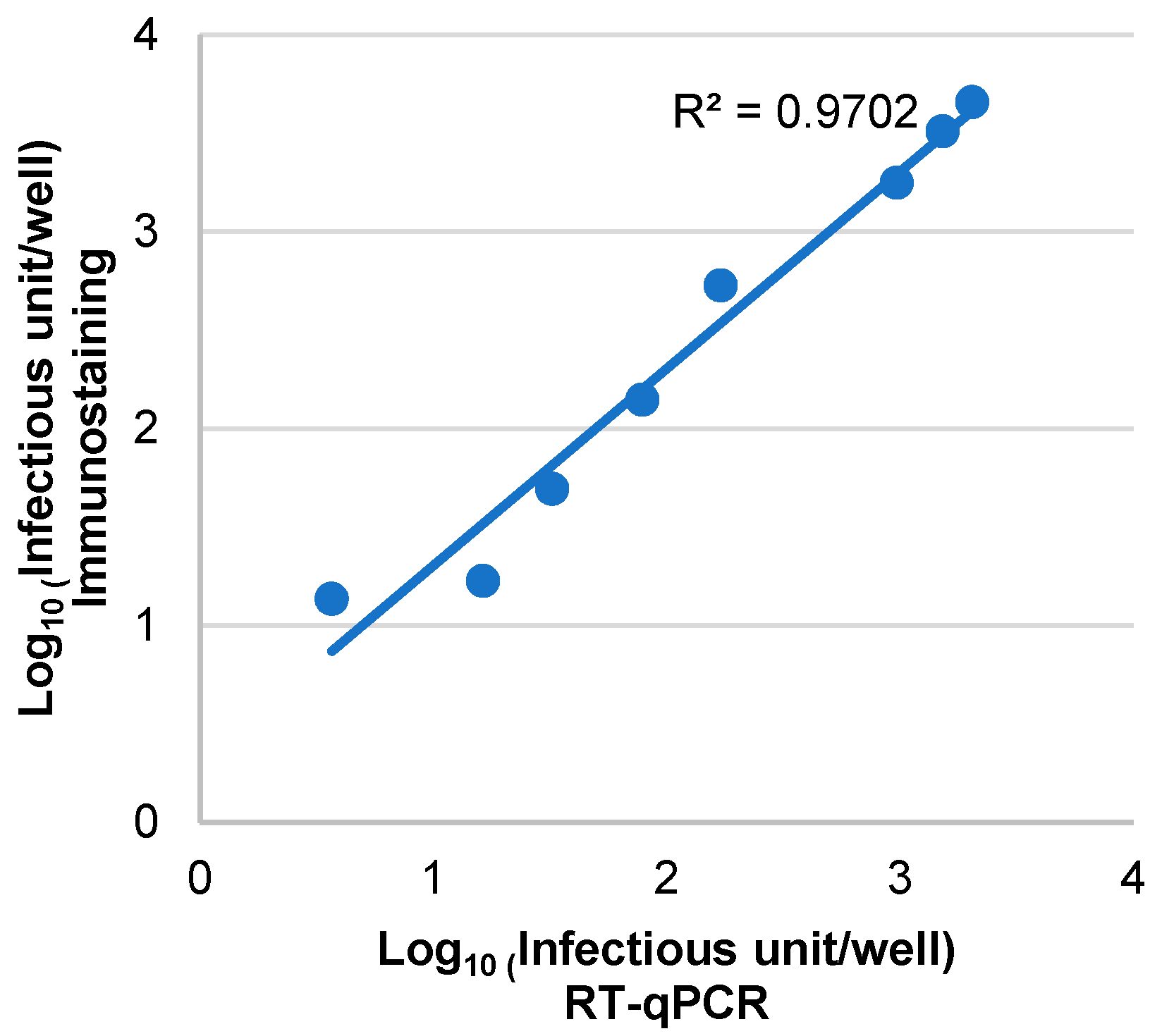
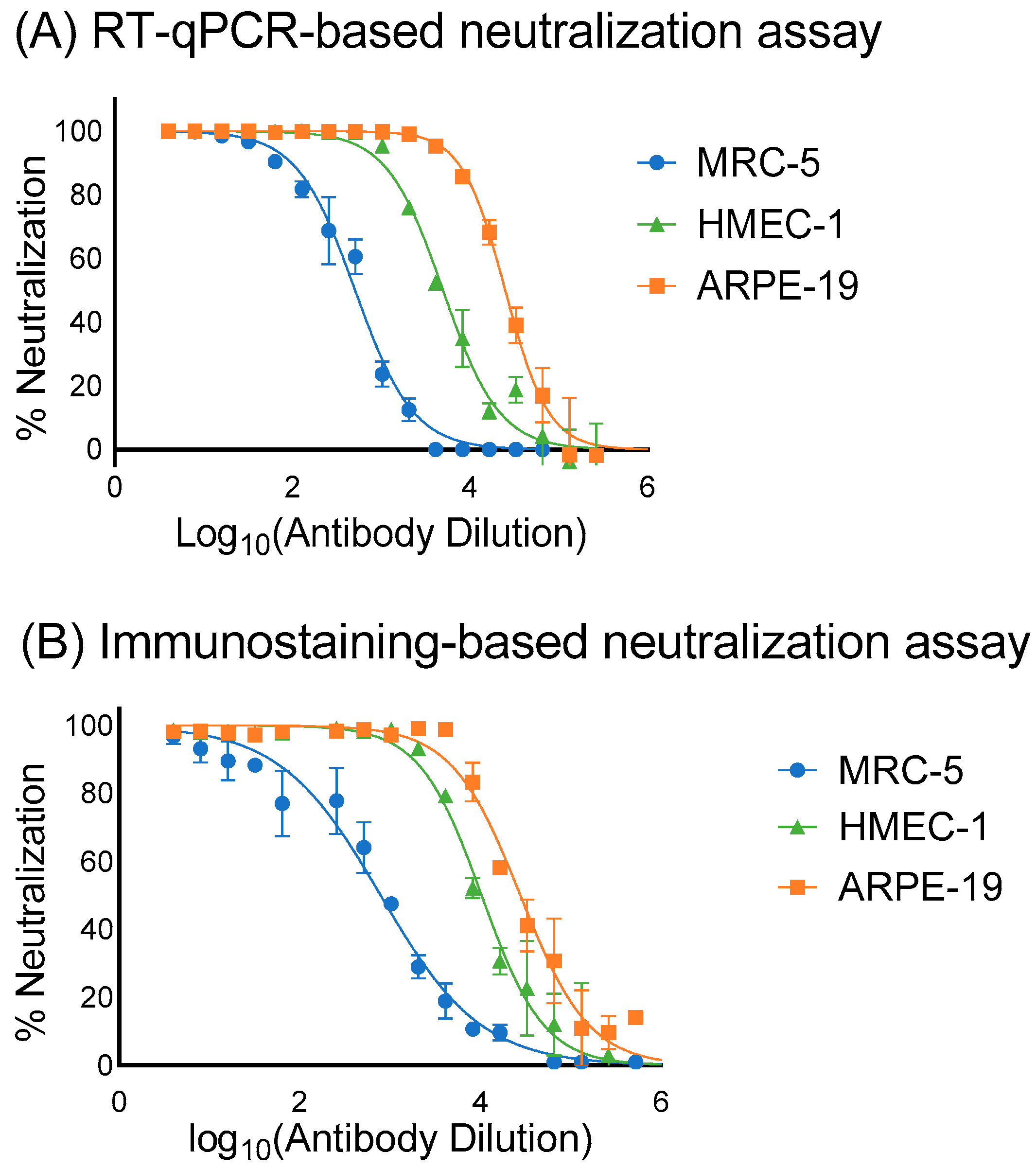
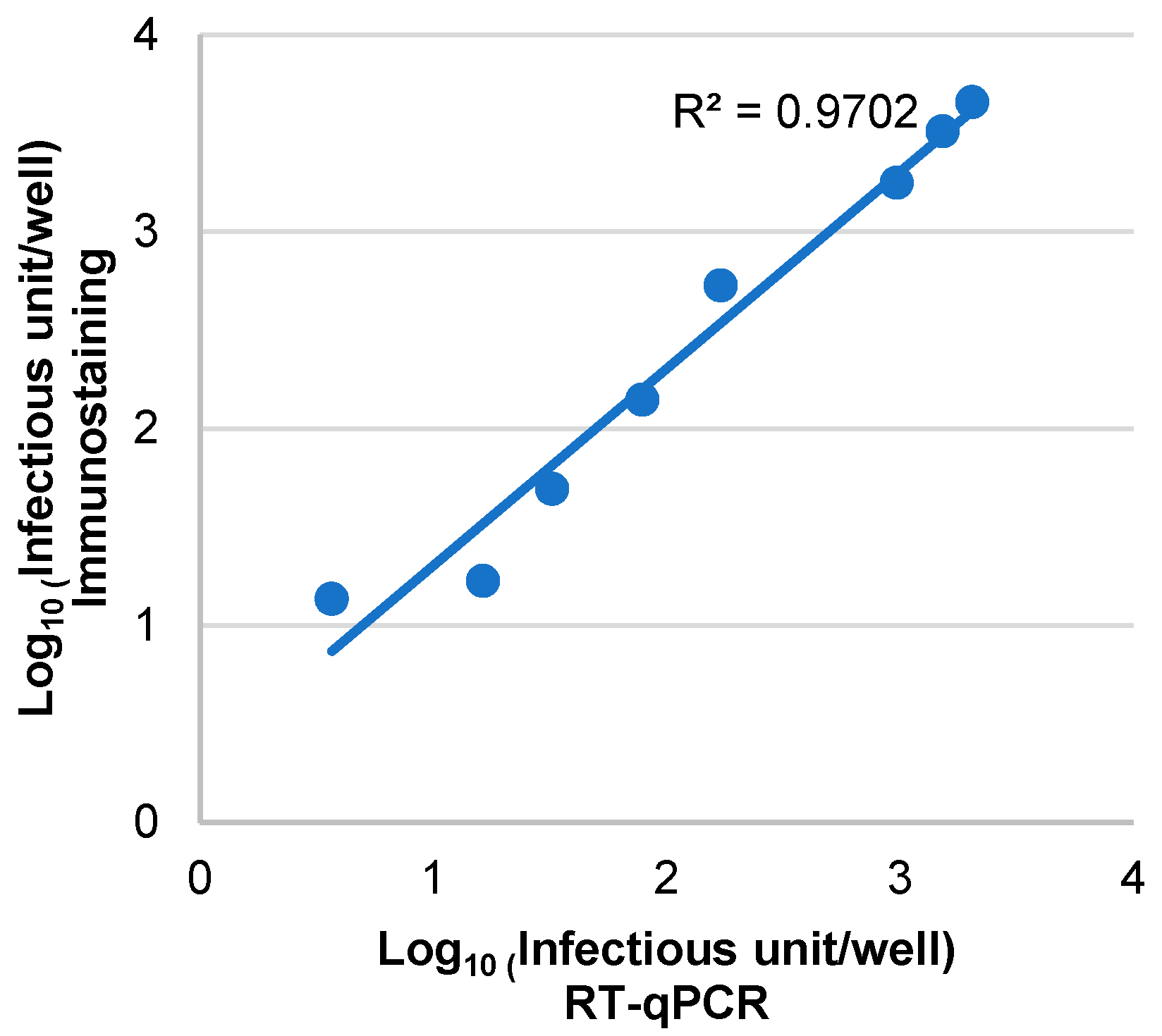
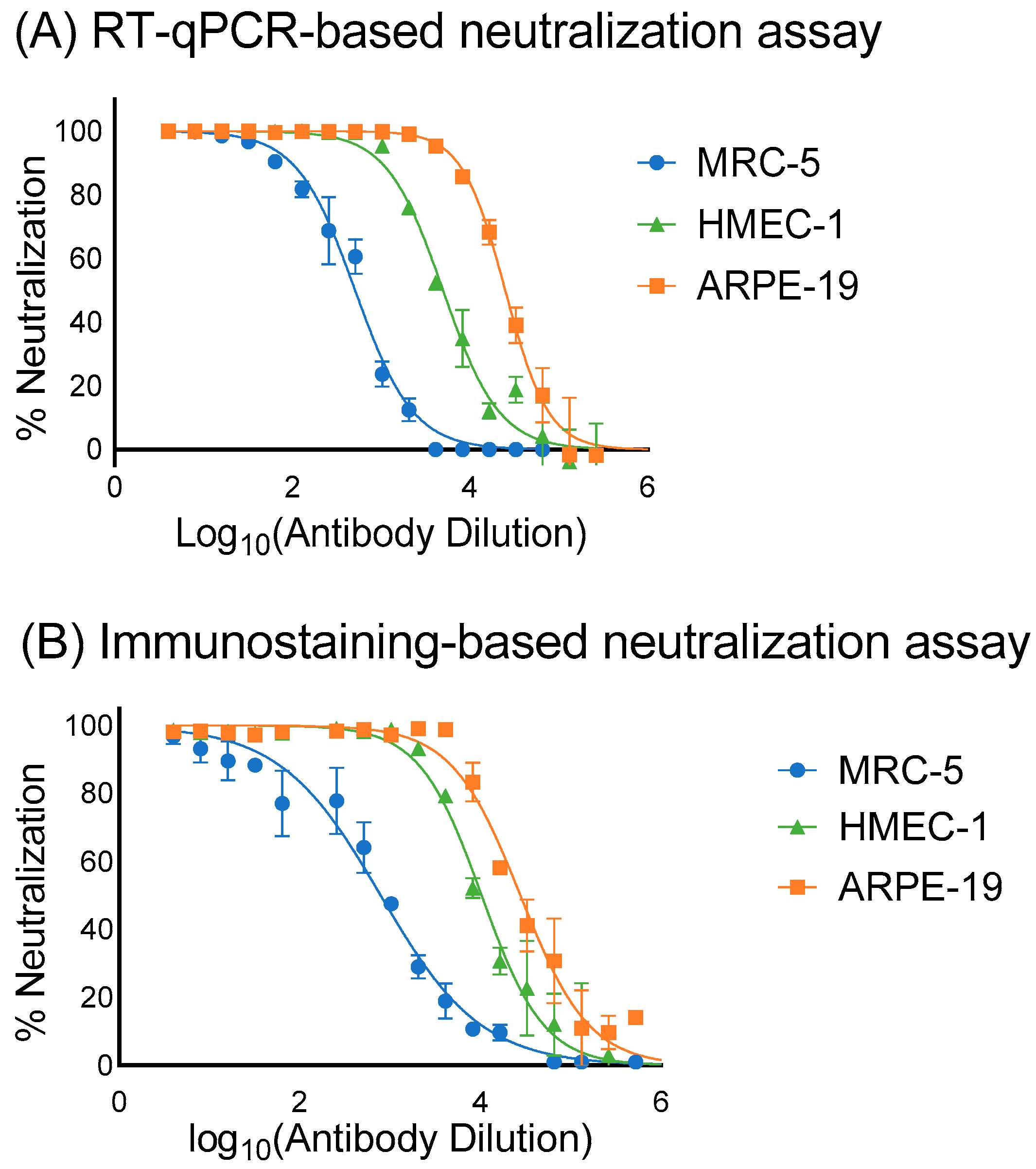
3.3. Assessment of the Sensitivity, Specificity and Correlation of RT-qPCR and Immunostaining Neutralization Assays
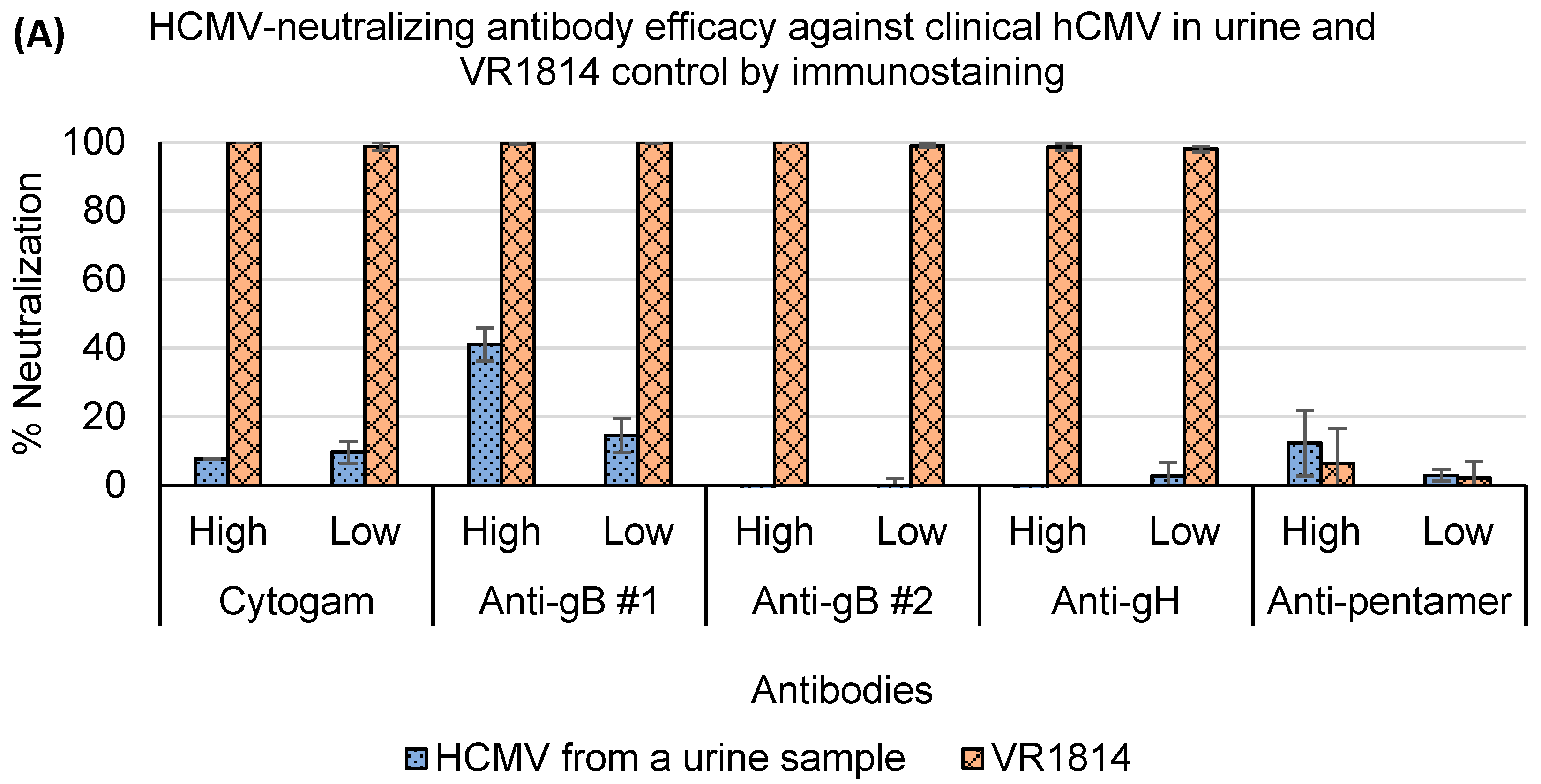
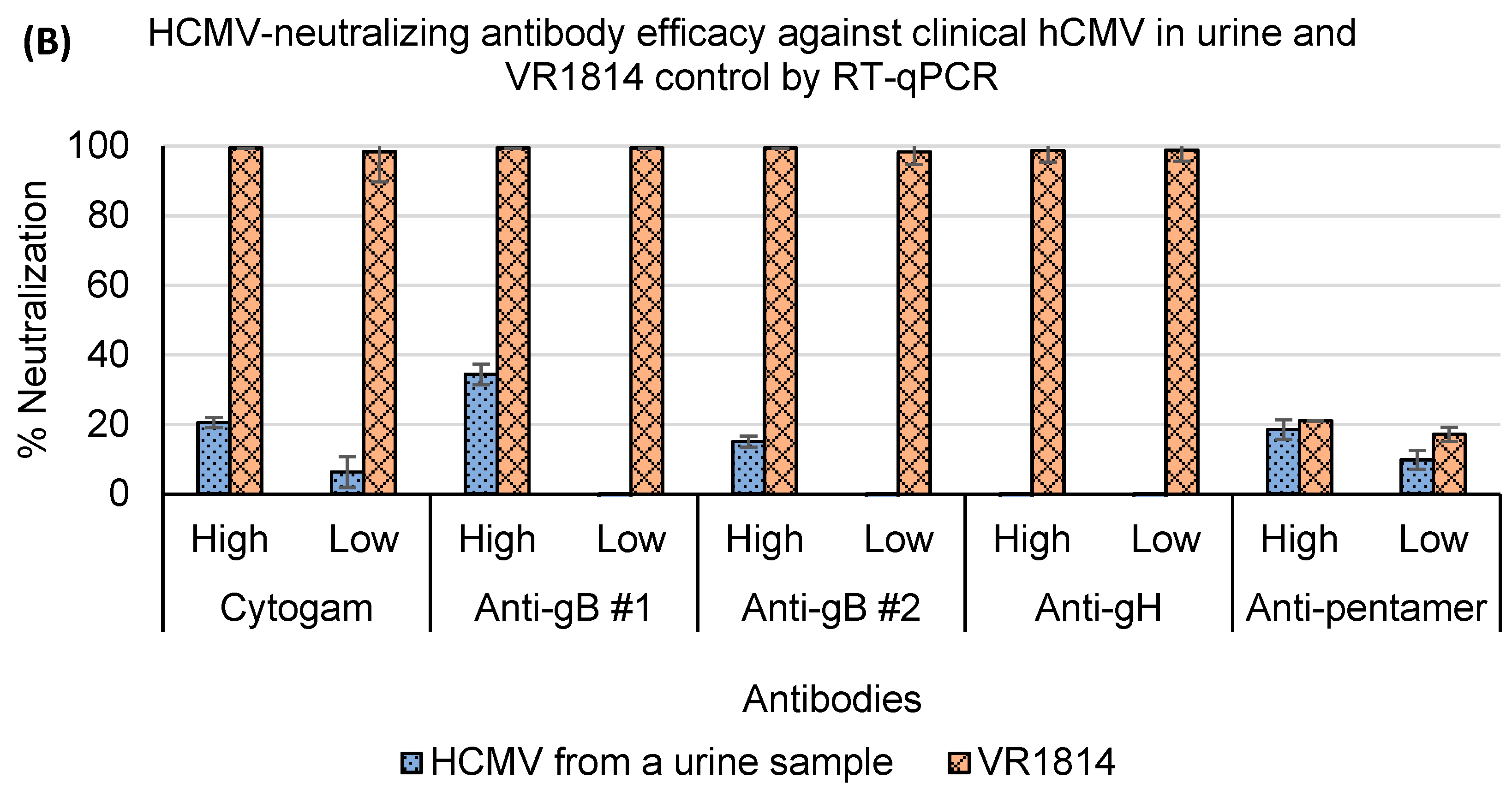
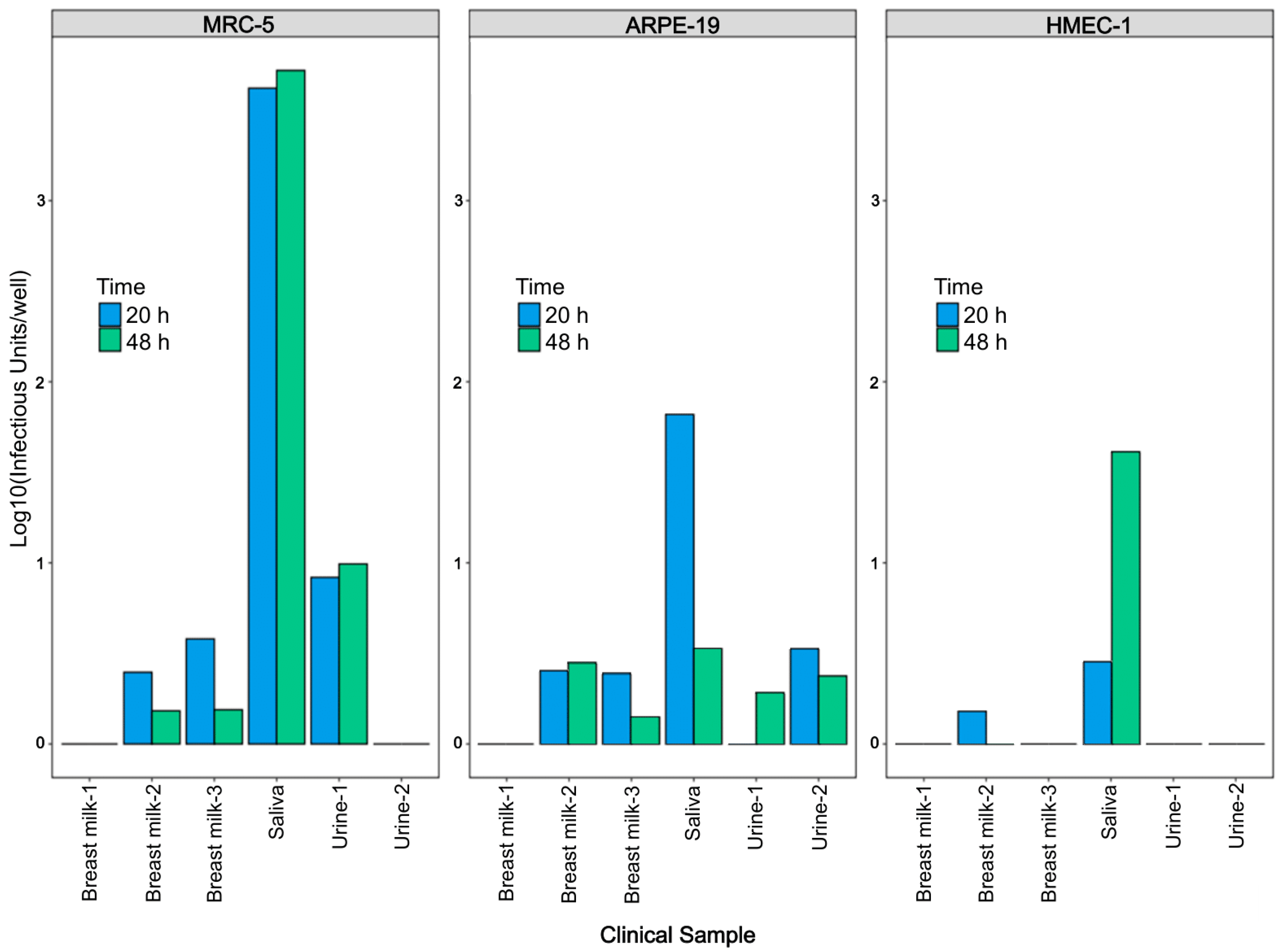
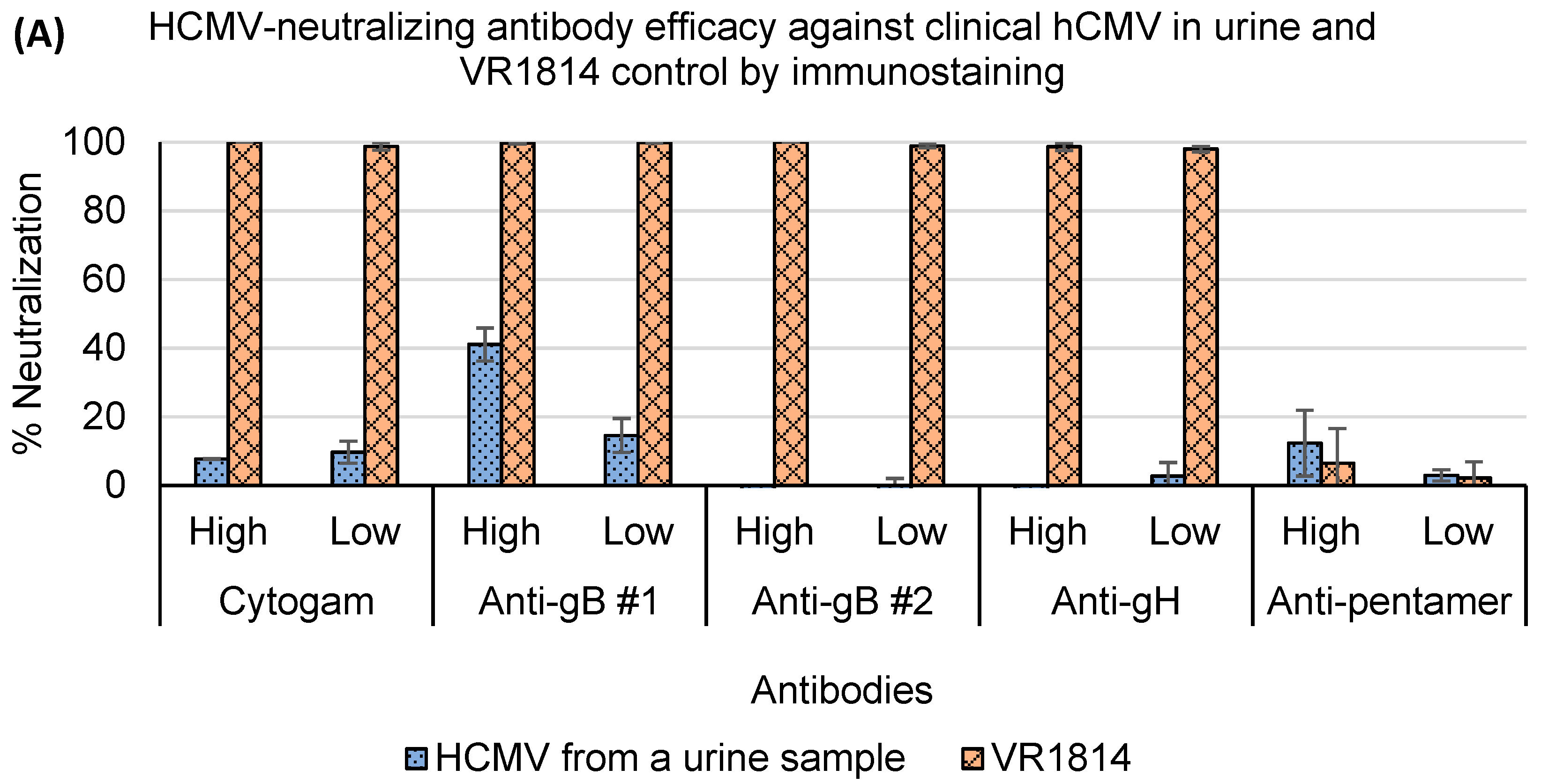
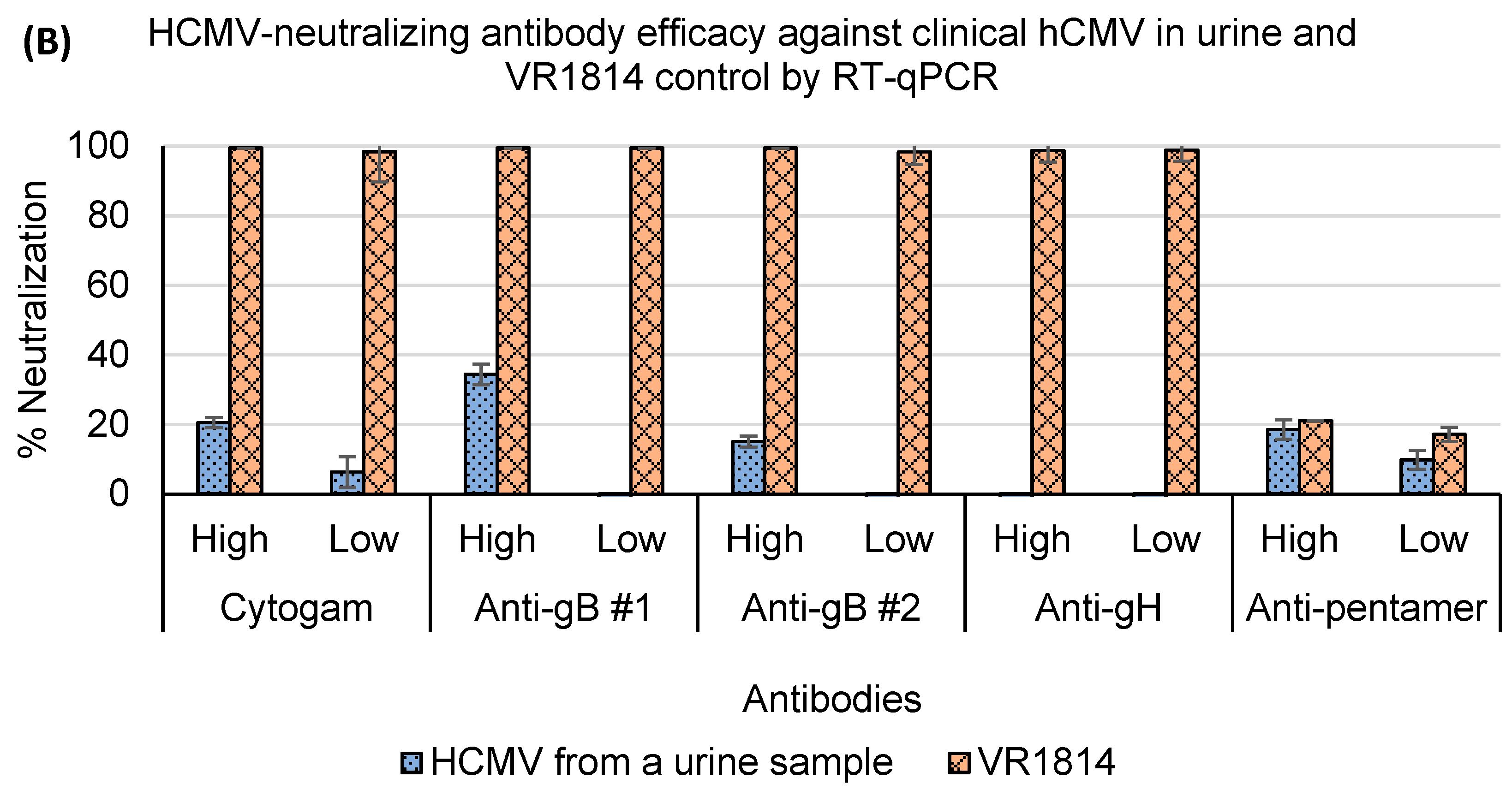
3.4. Assessment of Antibody Neutralization Activity against VR1814 and hCMV Strains in Primary Clinical Samples by RT-qPCR and Immunostaining Neutralization Assays
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American type culture collection |
| CV | Coefficient of variation |
| Ct | cycle threshold |
| CMV | Cytomegalovirus |
| DMEM | Dulbecco’s modified eagle’s medium |
| EGF | Epidermal growth factor |
| FBS | Fetal bovine serum |
| gB/H/L/O | Glycoprotein B/H/L/O |
| IC50 | Half maximal inhibitory concentration |
| h.p.i. | Hours post-infection |
| hCMV | Human cytomegalovirus |
| IE | Immediate-early |
| IgG | Immunoglobulin G |
| IU | Infectious units |
| MEM | Minimum essential medium eagle |
| PBS | Phosphate-buffered saline |
| PC | Pentameric complex |
| PCR | Polymerase chain reaction |
| rcf | relative centrifugal force |
| RT-qPCR | Reverse transcription-quantitative polymerase chain reaction |
| SD | Standard deviation |
References
- Louten, J. Herpesviruses. In Essential Human Virology; Academic Press: Cambridge, MA, USA, 2016; pp. 235–256. [Google Scholar]
- Goodrum, F.; Caviness, K.; Zagallo, P. Human Cytomegalovirus Persistence. Cell. Microbiol. 2012, 14, 644. [Google Scholar] [CrossRef]
- Bristow, B.N.; O’Keefe, K.A.; Shafir, S.C.; Sorvillo, F.J. Congenital Cytomegalovirus Mortality in the United States, 1990–2006. PLoS Negl. Trop. Dis. 2011, 5, e1140. [Google Scholar] [CrossRef]
- Manandhar, T.; Hò, G.G.T.; Pump, W.C.; Blasczyk, R.; Bade-Doeding, C. Battle between Host Immune Cellular Responses and Hcmv Immune Evasion. Int. J. Mol. Sci. 2019, 20, 3626. [Google Scholar] [CrossRef]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New Estimates of the Prevalence of Neurological and Sensory Sequelae and Mortality Associated with Congenital Cytomegalovirus Infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Cheung, T.W.; Teich, S.A. Cytomegalovirus Infection in Patients with HIV Infection. Mt. Sinai J. Med. 1999, 66, 113–124. [Google Scholar]
- Blanco-Lobo, P.; Bulnes-Ramos, Á.; McConnell, M.J.; Navarro, D.; Pérez-Romero, P. Applying Lessons Learned from Cytomegalovirus Infection in Transplant Patients to Vaccine Design. Drug Discov. Today 2016, 21, 674–681. [Google Scholar] [CrossRef]
- Stratton, K.R.; Durch, J.S.; Lawrence, R.S. Vaccines for the 21st Century; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.-L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine Prevention of Maternal Cytomegalovirus Infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Munoz, F.M.; Callahan, S.T.; Rupp, R.; Wootton, S.H.; Edwards, K.M.; Turley, C.B.; Stanberry, L.R.; Patel, S.M.; Mcneal, M.M.; et al. Safety and Efficacy of a Cytomegalovirus Glycoprotein B (GB) Vaccine in Adolescent Girls: A Randomized Clinical Trial. Vaccine 2016, 34, 313–319. [Google Scholar] [CrossRef]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.H.; Hauw The, T.; Jahn, G. Fibroblasts, Epithelial Cells, Endothelial Cells and Smooth Muscle Cells Are Major Targets of Human Cytomegalovirus Infection in Lung and Gastrointestinal Tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef]
- Nguyen, C.; Kamil, J. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef]
- Li, F.; Freed, D.C.; Tang, A.; Rustandi, R.R.; Troutman, M.C.; Espeseth, A.S.; Zhang, N.; An, Z.; McVoy, M.; Zhu, H.; et al. Complement Enhances in Vitro Neutralizing Potency of Antibodies to Human Cytomegalovirus Glycoprotein B (GB) and Immune Sera Induced by GB/MF59 Vaccination. NPJ. Vaccines 2017, 2, 36. [Google Scholar] [CrossRef]
- Loughney, J.W.; Rustandi, R.R.; Wang, D.; Troutman, M.C.; Dick, L.W.; Li, G.; Liu, Z.; Li, F.; Freed, D.C.; Price, C.E.; et al. Soluble Human Cytomegalovirus GH/GL/PUL128–131 Pentameric Complex, but Not GH/GL, Inhibits Viral Entry to Epithelial Cells and Presents Dominant Native Neutralizing Epitopes. J. Biol. Chem. 2015, 290, 15985–15995. [Google Scholar] [CrossRef]
- Wang, W.-D.; Lee, G.-C.; Kim, Y.Y.; Lee, C.H. A Comparison between Low- and High-Passage Strains of Human Cytomegalovirus. J. Microbiol. Biotechnol. 2016, 26, 1800–1807. [Google Scholar] [CrossRef]
- Cui, X.; Adler, S.P.; Schleiss, M.R.; Arav-Boger, R.; Harrison, G.J.D.; McVoy, M.A. Cytomegalovirus Virions Shed in Urine Have a Reversible Block to Epithelial Cell Entry and Are Highly Resistant to Antibody Neutralization. Clin. Vaccine Immunol. 2017, 24, e00024-17. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human Cytomegalovirus Entry into Epithelial and Endothelial Cells Depends on Genes UL128 to UL150 and Occurs by Endocytosis and Low-PH Fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef]
- Wang, X.; Peden, K.; Murata, H. RT-QPCR-Based Microneutralization Assay for Human Cytomegalovirus Using Fibroblasts and Epithelial Cells. Vaccine 2015, 33, 7254–7261. [Google Scholar] [CrossRef]
- Wentworth, B.B.; French, L. Plaque Assay of Cytomegalovirus Strains of Human Origin. Exp. Biol. Med. 1970, 135, 253–258. [Google Scholar] [CrossRef]
- Shatzkes, K.; Teferedegne, B.; Murata, H. A Simple, Inexpensive Method for Preparing Cell Lysates Suitable for Downstream Reverse Transcription Quantitative PCR. Sci. Rep. 2014, 4, 4659. [Google Scholar] [CrossRef]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of Human Monoclonal Antibodies That Potently Neutralize Human Cytomegalovirus Infection by Targeting Different Epitopes on the GH/GL/UL128-131A Complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef]
- Abai, A.M.; Smith, L.R.; Wloch, M.K. Novel Microneutralization Assay for HCMV Using Automated Data Collection and Analysis. J. Immunol. Methods 2007, 322, 82–93. [Google Scholar] [CrossRef]
- Andersen, H.K. Cytomegalovirus Neutralization by Plaque Reduction. Arch. Gesamte Virusforsch. 1971, 35, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.J.; Dennis, J.; Lennette, E.H. Plaque Reduction Neutralization Test for Human Cytomegalovirus Based upon Enhanced Uptake of Neutral Red by Virus-Infected Cells. J. Clin. Microbiol. 1976, 4, 61–66. [Google Scholar] [CrossRef]
- Rabe, I.B.; Staples, J.E.; Villanueva, J.; Hummel, K.B.; Johnson, J.A.; Rose, L.; Hills, S.; Wasley, A.; Fischer, M.; Powers, A.M. Interim Guidance for Interpretation of Zika Virus Antibody Test Results. MMWR Morb. Mortal. Wkly. Rep. 2019, 65, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Landry, M.L.; Stanat, S.; Biron, K.; Brambilla, D.; Britt, W.; Jokela, J.; Chou, S.; Drew, W.L.; Erice, A.; Gilliam, B.; et al. A Standardized Plaque Reduction Assay for Determination of Drug Susceptibilities of Cytomegalovirus Clinical Isolates. Antimicrob. Agents Chemother. 2000, 44, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Harmenberg, J.; Brytting, M.; Jabs, D.A.; Forman, M.S.; Enger, C.L. Limitations of Cytomegalovirus Testing. Antimicrob. Agents Chemother. 1999, 43, 1528. [Google Scholar] [CrossRef]
- Khan, G.; Kangro, H.O.; Coates, P.J.; Heath, R.B. Inhibitory Effects of Urine on the Polymerase Chain Reaction for Cytomegalovirus DNA. J. Clin. Pathol. 1991, 44, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Ochert, A.S.; Boulter, A.W.; Birnbaum, W.; Johnson, N.W.; Teo, C.G. Inhibitory Effect of Salivary Fluids on PCR: Potency and Removal. Genome Res. 1994, 3, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A.; Hamed, S.S.; Zhang, L.K. Use of Different Parameters and Equations for Calculation of IC50 Values in Efflux Assays: Potential Sources of Variability in IC50 Determination. AAPS J. 2014, 16, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Paolini, G.V.; Lyons, R.A.; Laflin, P. How Desirable Are Your IC50s? A Way to Enhance Screening-Based Decision Making. J. Biomol. Screen. 2010, 15, 1183–1193. [Google Scholar] [CrossRef]
- ISIRV Antiviral Group. Analysis of IC50 Data. International Society for Influenza and Other Respiratory Virus Diseases. Available online: https://isirv.org/site/index.php/methodology/analysis-of-ic50-data (accessed on 22 November 2021).
- Degraeve, G.M.; Cooney, J.D.; Mcintyre, D.O.; Pollock, T.L.; Reichenbach, N.G.; Dean, J.H.; Marcus, M.D. Variability in the Performance of the Seven-Day Fathead Minnow (Pimephales Promelas) Larval Survival and Growth Test: An Intra- and Interlaboratory Study. Environ. Toxicol. Chem. 1991, 10, 1189–1203. [Google Scholar]
- Kalliokoski, T.; Kramer, C.; Vulpetti, A.; Gedeck, P. Comparability of Mixed IC50 Data—A Statistical Analysis. PLoS ONE 2013, 8, e61007. [Google Scholar] [CrossRef] [PubMed]
- Jabs, D.A.; Enger, C.; Forman, M.; Dunn, J.P. Incidence of Foscarnet Resistance and Cidofovir Resistance in Patients Treated for Cytomegalovirus Retinitis. Antimicrob. Agents Chemother. 1998, 42, 2240. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Coelingh, K.L.; Britt, W.J. Human Cytomegalovirus Neutralizing Antibody-Resistant Phenotype Is Associated with Reduced Expression of Glycoprotein H. J. Virol. 1995, 69, 6047–6053. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Schoppel, K.; Amvrossiadis, N.; Mach, M. Strain-Specific Neutralization of Human Cytomegalovirus Isolates by Human Sera. J. Virol. 1999, 73, 878–886. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hCMV Virions Input (log10 IU/Well *) | Mean Values of Crude Cell Lysate Supernatant/RNA Extraction (log10 IU/Well) | ||
|---|---|---|---|
| MRC-5 | ARPE-19 | HMEC-1 | |
| 4 | 5.01/5.01 | 5.17/4.68 | 5.05/4.91 |
| 3 | 3.58/3.50 | 4.53/4.32 | 4.57/4.20 |
| 2 | 2.61/2.36 | 2.82/2.73 | 2.90/2.69 |
| 1 | 1.26/1.12 | 2.17/2.08 | 2.09/1.73 |
| IU/Reaction | No. of Samples | SD | %CV |
|---|---|---|---|
| 1.1 × 104 | 9 | 0.04 | 0.94 |
| 2.6 × 103 | 9 | 0.16 | 4.56 |
| 4.3 × 102 | 9 | 0.07 | 2.48 |
| 3.0 × 102 | 9 | 0.11 | 4.25 |
| 2.9 × 102 | 9 | 0.19 | 7.87 |
| 35 | 9 | 0.08 | 5.32 |
| 10 | 9 | 0.08 | 7.40 |
| 5 | 9 | 0.10 | 13.31 |
| 5 | 9 | 0.11 | 15.08 |
| hCMV Input (IU/Well) | RT-qPCR-Based | Immunostaining-Based | ||
|---|---|---|---|---|
| Mean ± SD (IU/Well) | No. Pos | Mean ± SD (Viral Particles/Well) | No. Pos | |
| 20 | 22.47 ± 3.58 | 6/6 | 4 ± 2.76 | 6/6 |
| 10 | 10.1 ± 5.99 | 6/6 | 0.67 ± 1.03 | 2/6 |
| 1 | 6.55 ± 5.1 | 6/6 | 0 | 0/6 |
| Neg controls | - | 0/10 | 0.4 ± 0.7 | 3/10 |
| Cell Type | IC50 (μg/mL) | |
|---|---|---|
| RT-qPCR | Immunostaining | |
| MRC-5 | 393.31 | 254.36 |
| ARPE-19 | 8.04 | 7.23 |
| HMEC-1 | 40.3 | 19.4 |
| RT-qPCR | |||
| Positive | Negative | ||
| Immunostaining | Positive | 24 | 0 |
| Negative | 19 | 35 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Hasing, M.E.; Preiksaitis, J.K.; Pang, X. Evaluation of a Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)-Based Microneutralization Assay for Assessing Clinical Human Cytomegalovirus-Neutralizing Antibody Activity. Microorganisms 2024, 12, 742. https://doi.org/10.3390/microorganisms12040742
Yu J, Hasing ME, Preiksaitis JK, Pang X. Evaluation of a Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)-Based Microneutralization Assay for Assessing Clinical Human Cytomegalovirus-Neutralizing Antibody Activity. Microorganisms. 2024; 12(4):742. https://doi.org/10.3390/microorganisms12040742
Chicago/Turabian StyleYu, Jiaao, Maria E. Hasing, Jutta K. Preiksaitis, and Xiaoli Pang. 2024. "Evaluation of a Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)-Based Microneutralization Assay for Assessing Clinical Human Cytomegalovirus-Neutralizing Antibody Activity" Microorganisms 12, no. 4: 742. https://doi.org/10.3390/microorganisms12040742