
Deciphering Microbial Shifts in the Gut and Lung Microbiomes of COVID-19 Patients
Abstract
:1. Introduction
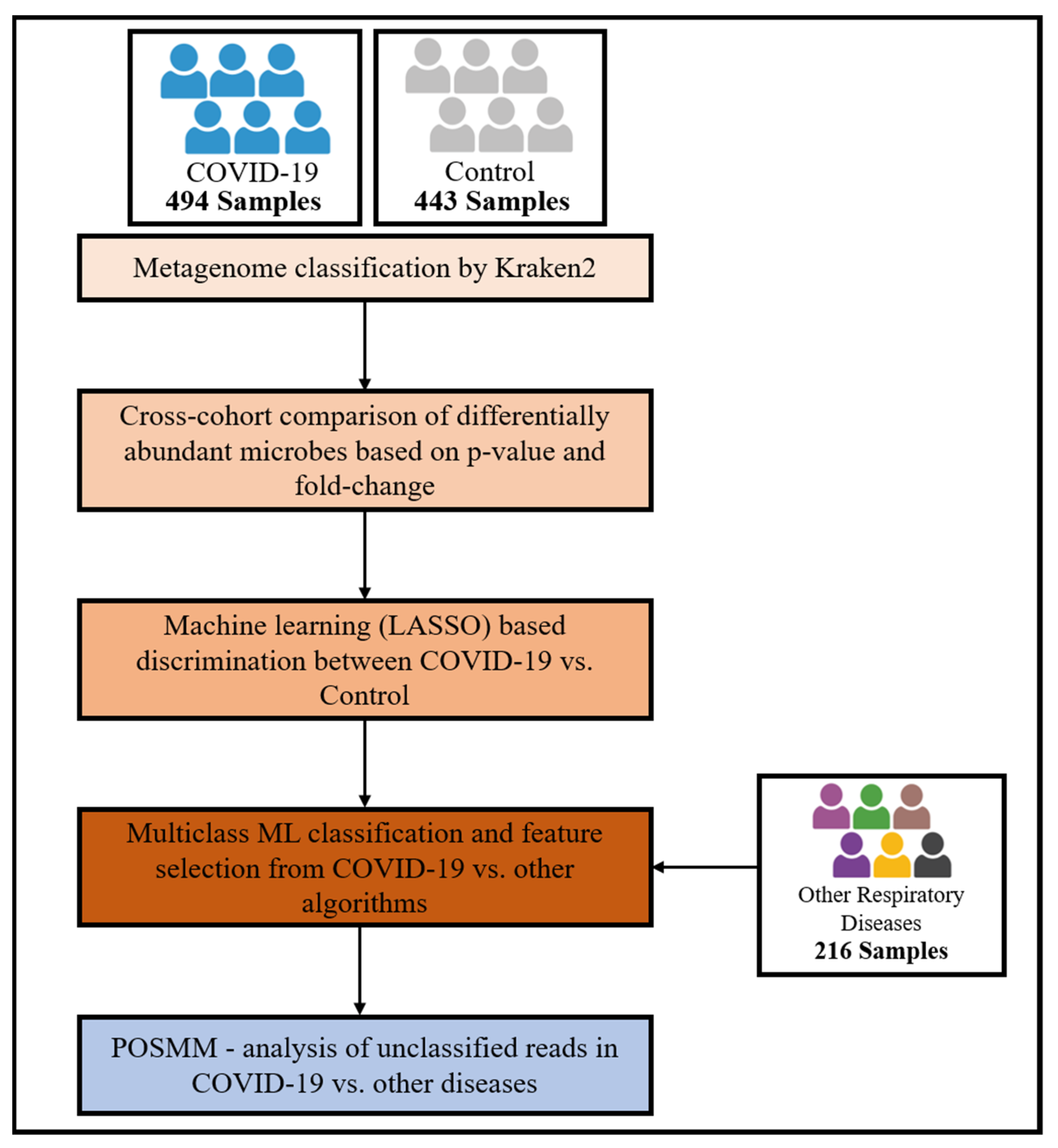
2. Materials and Methods
2.1. Metagenomic Data
2.2. Taxonomic Classification
2.3. Biomarker Identification
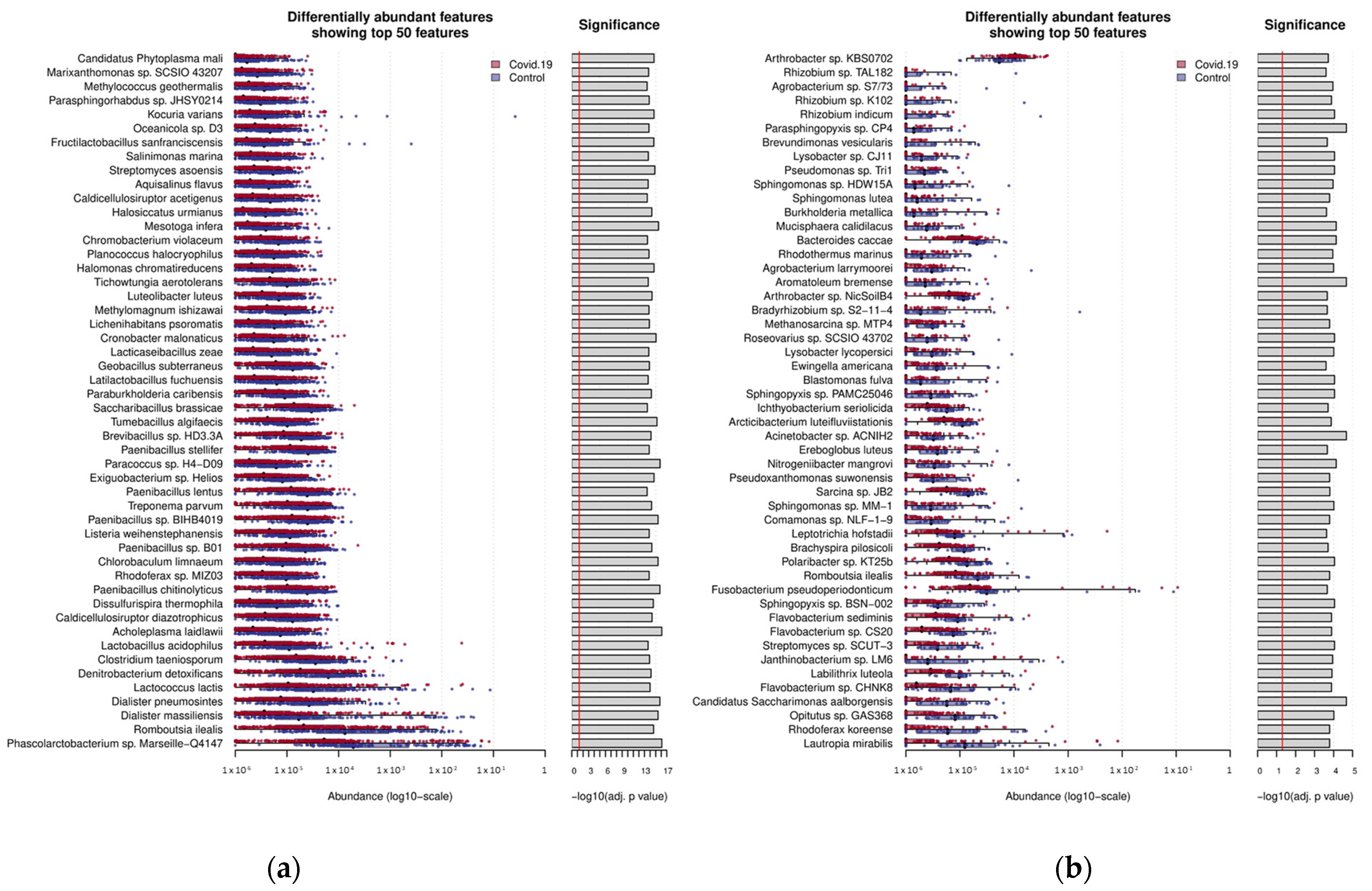
2.4. Differential Abundance Estimation for Classified Reads
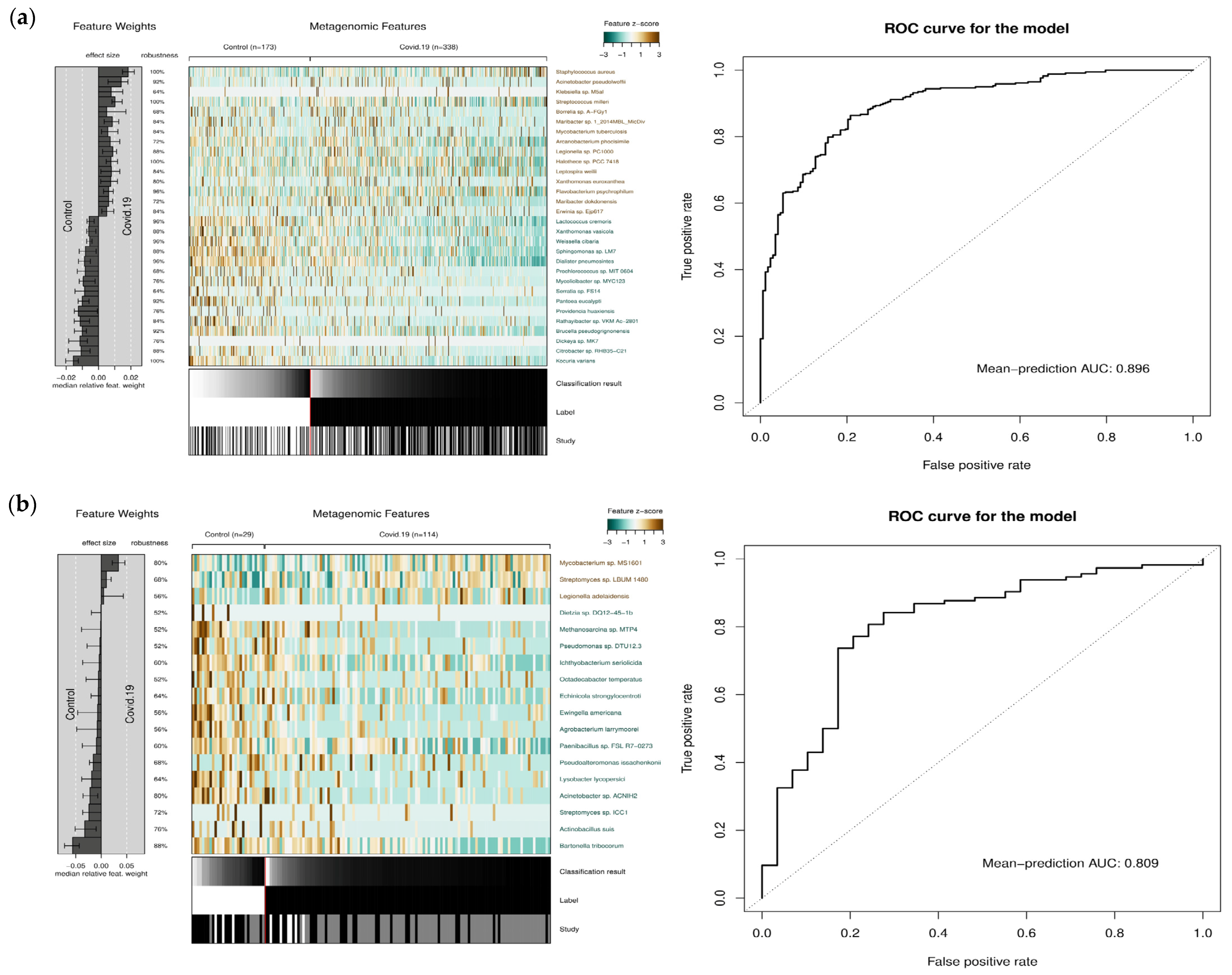
2.5. Binary Machine Learning Analysis
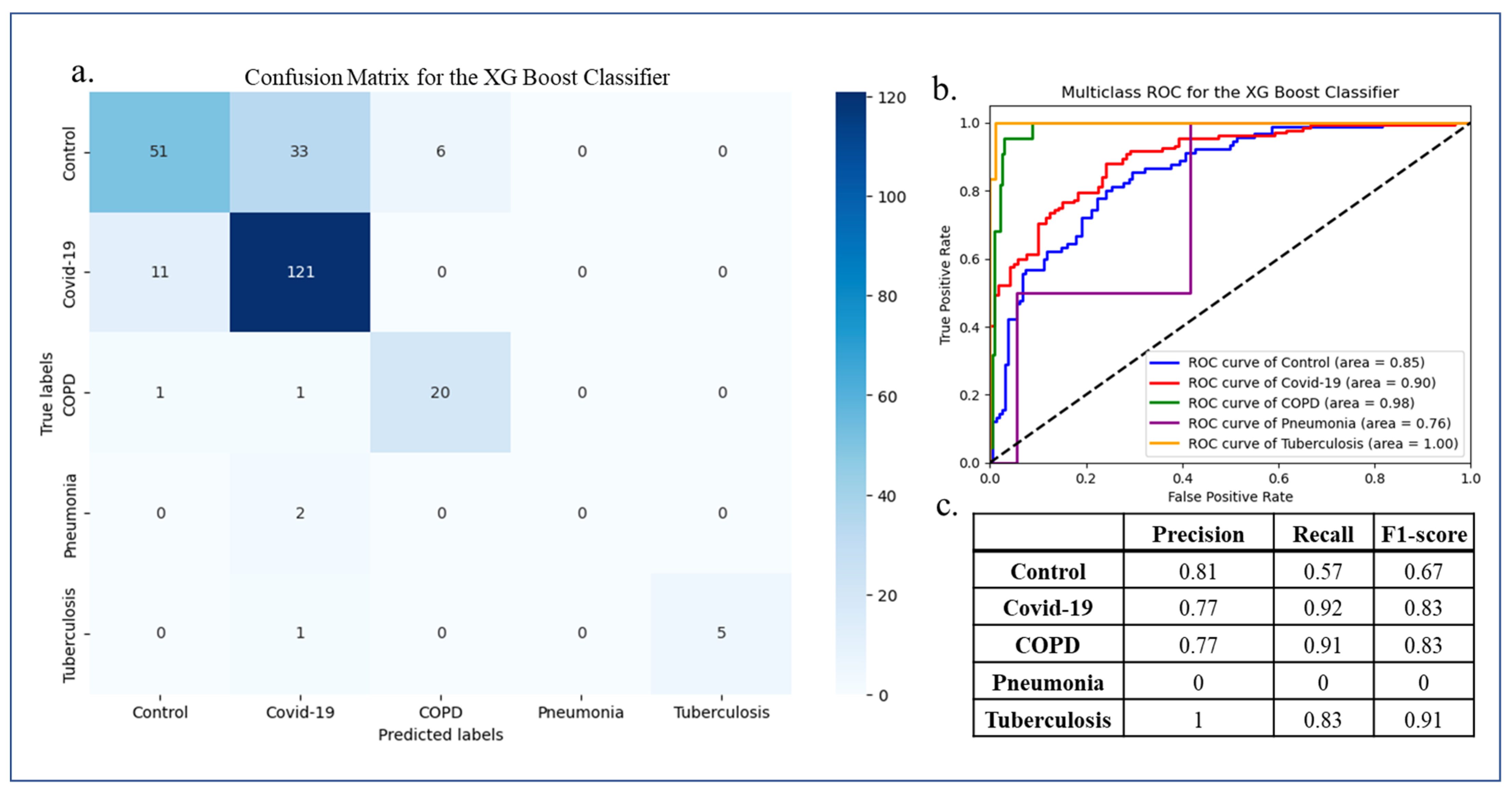
2.6. Multiclass Machine Learning Classification
3. Results
3.1. A Meta-Analysis of COVID-19 and Other Respiratory Diseases’ Gut and Lung Microbiome Datasets
3.2. Classification of COVID-19 and Control Microbiome Samples Based on Microbial Abundance Using Machine Learning
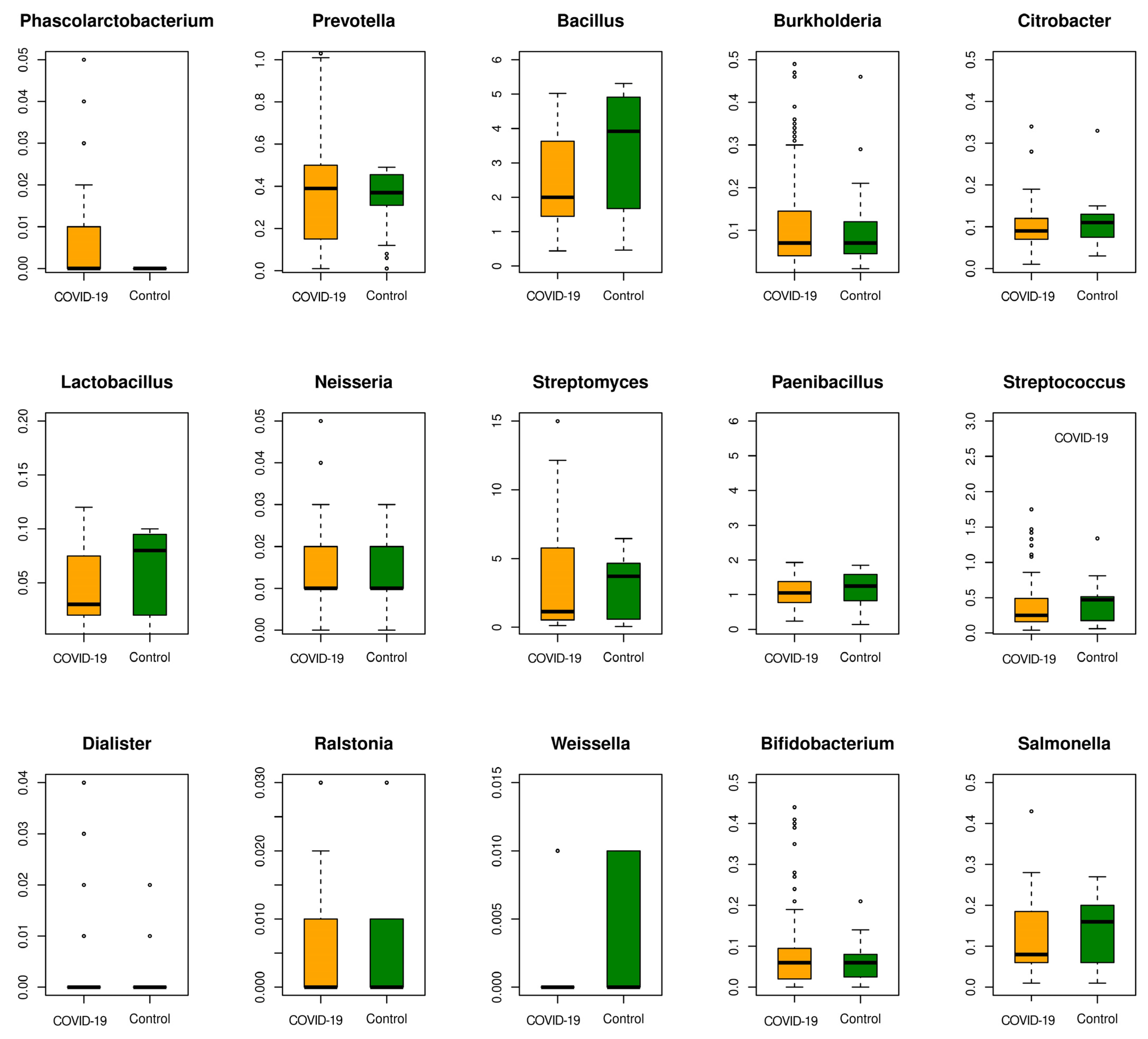
3.3. Multi-Disease Comparison of Differentially Associated Microbes
3.4. Multiclass Machine Learning Classification
3.5. Classifying the “Unclassified” Microbiome Reads Using POSMM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Marin, I.A.; Goertz, J.E.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota alteration is associated with the development of stress-induced despair behavior. Sci. Rep. 2017, 7, 43859. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef] [PubMed]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: Estimation and application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Qiu, Y.; He, J.-S.; Tan, J.-Y.; Li, X.-H.; Liang, J.; Shen, J.; Zhu, L.-R.; Chen, Y.; Iacucci, M.; et al. Manifestations and prognosis of gastrointestinal and liver involvement in patients with COVID-19: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 667–678. [Google Scholar] [PubMed]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.Y.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Reuben, R.C.; Beugnon, R.; Jurburg, S.D. COVID-19 alters human microbiomes: A meta-analysis. Front. Cell. Infect. Microbiol. 2023, 13, 1211348. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Okin, D.; Drew, D.A.; Battista, V.M.; Jesudasen, S.J.; Kuntz, T.M.; Bhosle, A.; Thompson, K.N.; Reinicke, T.; Lo, C.-H.; et al. Metagenomic assessment of gut microbial communities and risk of severe COVID-19. Genome Med. 2023, 15, 49. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, Y.; Li, Y.; Wu, Q.; Wu, J.; Park, S.-K.; Guo, C.; Lu, J. Meta-analysis of 16S rRNA microbial data identified alterations of the gut microbiota in COVID-19 patients during the acute and recovery phases. BMC Microbiol. 2022, 22, 274. [Google Scholar] [CrossRef]
- Li, J.; Ghosh, T.S.; McCann, R.; Mallon, P.; Hill, C.; Draper, L.; Schult, D.; Fanning, L.J.; Shannon, R.; Sadlier, C.; et al. Robust cross-cohort gut microbiome associations with COVID-19 severity. Gut Microbes 2023, 15, 2242615. [Google Scholar] [CrossRef]
- Burks, D.J.; Pusadkar, V.; Azad, R.K. POSMM: An efficient alignment-free metagenomic profiler that complements alignment-based profiling. Environ. Microbiome 2023, 18, 16. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2020, 36, 1303–1304. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, R.; Rohini, R. LASSO: A feature selection technique in predictive modeling for machine learning. In Proceedings of the 2016 IEEE International Conference on Advances in Computer Applications, ICACA 2016, Coimbatore, India, 24 October 2016; pp. 18–20. [Google Scholar] [CrossRef]
- Wirbel, J.; Zych, K.; Essex, M.; Karcher, N.; Kartal, E.; Salazar, G.; Bork, P.; Sunagawa, S.; Zeller, G. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021, 22, 93. [Google Scholar] [CrossRef]
- Zeng, X.; Martinez, T.R. Feature weighting using neural networks. In Proceedings of the IEEE International Conference on Neural Networks—Conference Proceedings, Budapest, Hungary, 25–29 July 2004; Volume 2, pp. 1327–1330. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Nguyen, Q.V.; Chong, L.C.; Hor, Y.-Y.; Lew, L.-C.; Rather, I.A.; Choi, S.-B. Role of Probiotics in the Management of COVID-19: A Computational Perspective. Nutrients 2022, 14, 274. [Google Scholar] [CrossRef]
- Kim, J.G.; Zhang, A.; Rauseo, A.M.; Goss, C.W.; Mudd, P.A.; O’Halloran, J.A.; Wang, L. The salivary and nasopharyngeal microbiomes are associated with SARS-CoV-2 infection and disease severity. J. Med. Virol. 2023, 95, e28445. [Google Scholar] [CrossRef]
- Wu, F.; Guo, X.; Zhang, J.; Zhang, M.; Ou, Z.; Peng, Y. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp. Ther. Med. 2017, 14, 3122–3126. [Google Scholar] [CrossRef]
- Gerritsen, J.; Hornung, B.; Renckens, B.; van Hijum, S.A.; Dos Santos, V.A.M.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. Genomic and functional analysis of Romboutsia ilealis CRIBT reveals adaptation to the small intestine. PeerJ 2017, 2017, e3698. [Google Scholar] [CrossRef]
- Ghattargi, V.C.; Nimonkar, Y.S.; Sape, K.; Prakash, O.; Suryavanshi, M.V.; Shouche, Y.S.; Meti, B.S.; Pawar, S.P. Functional and Comparative Genomics of Niche-Specific Adapted Actinomycetes Kocuria rhizophila Strain D2 Isolated from Healthy Human Gut. bioRxiv 2018. [Google Scholar] [CrossRef]
- Villoslada-Blanco, P.; Pérez-Matute, P.; Recio-Fernández, E.; Íñiguez, M.; Blanco-Navarrete, P.; Metola, L.; Ibarra, V.; Alba, J.; de Toro, M.; Oteo, J.A. Beyond the effects of HIV infection and integrase inhibitors-based therapies on oral bacteriome. Sci. Rep. 2023, 13, 14327. [Google Scholar] [CrossRef]
- Zafar, H.; Saier, M.H. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Delday, M.; Mulder, I.; Logan, E.T.; Grant, G. Bacteroides thetaiotaomicron Ameliorates Colon Inflammation in Preclinical Models of Crohn’s Disease. Inflamm. Bowel Dis. 2019, 25, 85–96. [Google Scholar] [CrossRef]
- Becker, H.E.F.; Jamin, C.; Bervoets, L.; Boleij, A.; Xu, P.; Pierik, M.J.; Stassen, F.R.M.; Savelkoul, P.H.M.; Penders, J.; Jonkers, D.M.A.E. Higher Prevalence of Bacteroides fragilis in Crohn’s Disease Exacerbations and Strain-Dependent Increase of Epithelial Resistance. Front. Microbiol. 2021, 12, 598232. [Google Scholar] [CrossRef]
- Ezeji, J.C.; Sarikonda, D.K.; Hopperton, A.; Erkkila, H.L.; Cohen, D.E.; Martinez, S.P.; Cominelli, F.; Kuwahara, T.; Dichosa, A.E.K.; Good, C.E.; et al. Parabacteroides distasonis: Intriguing aerotolerant gut anaerobe with emerging antimicrobial resistance and pathogenic and probiotic roles in human health. Gut Microbes 2021, 13, 1922241. [Google Scholar] [CrossRef]
- Nagayama, M.; Yano, T.; Atarashi, K.; Tanoue, T.; Sekiya, M.; Kobayashi, Y.; Sakamoto, H.; Miura, K.; Sunada, K.; Kawaguchi, T.; et al. TH1 cell-inducing Escherichia coli strain identified from the small intestinal mucosa of patients with Crohn’s disease. Gut Microbes 2020, 12, 1788898. [Google Scholar] [CrossRef] [PubMed]
- Zabetakis, I.; Lordan, R.; Norton, C.; Tsoupras, A. COVID-19: The Inflammation Link and the Role of Nutrition in Potential Mitigation. Nutrients 2020, 12, 1466. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Horn, K.J.; Schopper, M.A.; Drigot, Z.G.; Clark, S.E. Airway Prevotella promote TLR2-dependent neutrophil activation and rapid clearance of Streptococcus pneumoniae from the lung. Nat. Commun. 2022, 13, 3321. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhao, Y. Comparative resistomic analyses of Lysobacter species with high intrinsic multidrug resistance. J. Glob. Antimicrob. Resist. 2019, 19, 320–327. [Google Scholar] [CrossRef]
- Sencio, V.; Machado, M.G.; Trottein, F. The lung–gut axis during viral respiratory infections: The impact of gut dysbiosis on secondary disease outcomes. Mucosal Immunol. 2021, 14, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gu, S.; Gong, Y.; Li, B.; Lu, H.; Li, Q.; Zhang, R.; Gao, X.; Wu, Z.; Zhang, J.; et al. Clinical Significance of the Correlation between Changes in the Major Intestinal Bacteria Species and COVID-19 Severity. Engineering 2020, 6, 1178–1184. [Google Scholar] [CrossRef]
- Verhasselt, H.L.; Buer, J.; Dedy, J.; Ziegler, R.; Steinmann, J.; Herbstreit, F.; Brenner, T.; Rath, P.M. COVID-19 Co-infection with Legionella pneumophila in 2 Tertiary-Care Hospitals, Germany. Emerg. Infect. Dis. 2021, 27, 1535. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, H.; Zhou, Q.; Feng, R.; Zhai, B. Impact of COVID-19 control measures on Legionella pneumophila infections in children in Henan, China. J. Infect. 2023, 87, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Nariya, S.; Harris, J.M.; Lynch, S.V.; Choy, D.F.; Arron, J.R.; Boushey, H. The airway microbiome in patients with severe asthma: Associations with disease features and severity. J. Allergy Clin. Immunol. 2015, 136, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Srinivasan, M.; Ghosh, T.S.; Mande, S.S. Xenobiotic Metabolism and Gut Microbiomes. PLoS ONE 2016, 11, e0163099. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Pramanik, S. Structural diversity, functional aspects and future therapeutic applications of human gut microbiome. Arch. Microbiol. 2021, 203, 5281–5308. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G.; Fischbach, M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414. [Google Scholar] [CrossRef]
- Yu, H.S.; Lee, N.K.; Choi, A.J.; Choe, J.S.; Bae, C.H.; Paik, H.D. Anti-Inflammatory Potential of Probiotic Strain Weissella cibaria JW15 Isolated from Kimchi through Regulation of NF-κB and MAPKs Pathways in LPS-Induced RAW 264.7 Cells. J. Microbiol. Biotechnol. 2019, 29, 1022–1032. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Discovery Datasets | BioProject ID | Number of Samples | Number of Diseased Samples | Number of Control Samples | Source | Year | Country | |
|---|---|---|---|---|---|---|---|---|
| COVID-19 | 1 | PRJDB13214 | 208 | 103 | 105 | Gut microbiome | 2023 | Japan |
| 2 | PRJNA781460 | 57 | 39 | 19 | Respiratory microbiome | 2021 | Sweden | |
| 3 | PRJNA656660 | 9 | 6 | 3 | Respiratory microbiome | 2020 | China | |
| 4 | PRJNA743981 | 99 | 79 | 20 | Respiratory microbiome | 2021 | USA | |
| 5 | PRJNA624223 | 50 | 15 | 15 | Gut microbiome | 2020 | China | |
| 6 | PRJNA650244 | 240 | 187 | 53 | Gut microbiome | 2020 | China | |
| Other respiratory diseases | ||||||||
| Pneumonia | 1 | PRJNA624223 | 21 | 6 | 15 | Gut microbiome | 2020 | China |
| COPD | 2 | PRJNA562766 | 57 | 29 | 28 | Gut microbiome | 2020 | Australia |
| COPD | 3 | PRJNA852674 | 135 | 99 | 36 | Respiratory microbiome | 2022 | China |
| Pulmonary tuberculosis | 4 | PRJNA401385 | 77 | 46 | 31 | Gut microbiome | 2017 | China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pusadkar, V.; Mazumder, A.; Azad, A.; Patil, D.; Azad, R.K. Deciphering Microbial Shifts in the Gut and Lung Microbiomes of COVID-19 Patients. Microorganisms 2024, 12, 1058. https://doi.org/10.3390/microorganisms12061058
Pusadkar V, Mazumder A, Azad A, Patil D, Azad RK. Deciphering Microbial Shifts in the Gut and Lung Microbiomes of COVID-19 Patients. Microorganisms. 2024; 12(6):1058. https://doi.org/10.3390/microorganisms12061058
Chicago/Turabian StylePusadkar, Vaidehi, Anirudh Mazumder, Abhijay Azad, Deepti Patil, and Rajeev K. Azad. 2024. "Deciphering Microbial Shifts in the Gut and Lung Microbiomes of COVID-19 Patients" Microorganisms 12, no. 6: 1058. https://doi.org/10.3390/microorganisms12061058