
Druggability Analysis of Protein Targets for Drug Discovery to Combat Listeria monocytogenes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Reference Protein Selection to Build the Druggability Analysis Model
2.1.2. Physical Characterization of Protein Structures
2.1.3. Target Protein Selection for Druggability Analysis
2.2. Methods
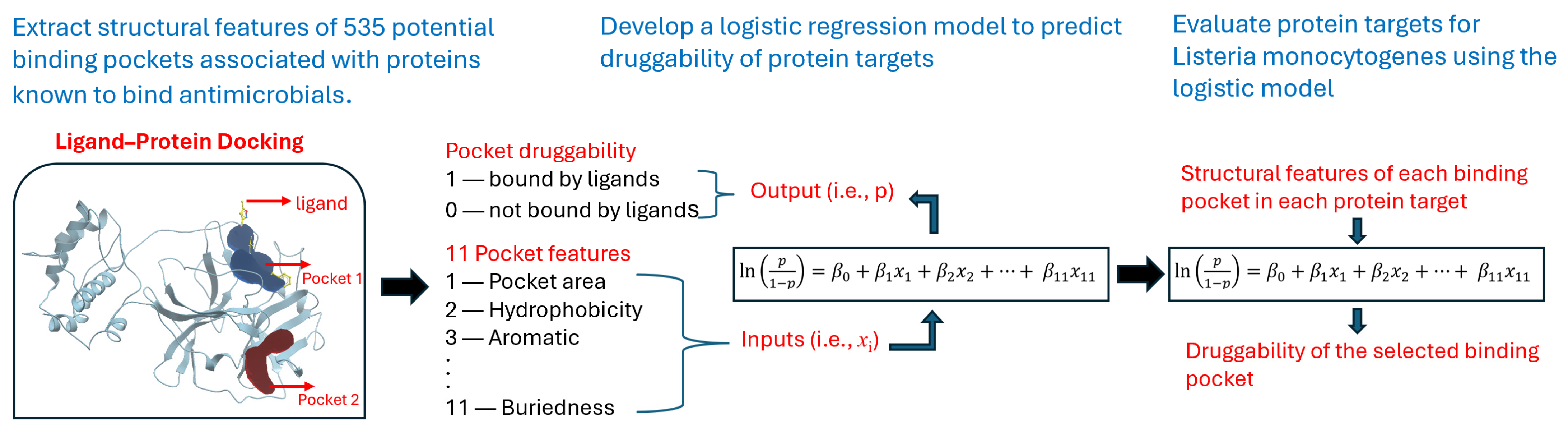
2.2.1. Statistical Analysis of Pocket Data for Reference Proteins
2.2.2. Preparation of the Reference Data Set
2.2.3. Logistic Regression
2.2.4. Analysis of the Target Data Set
3. Results
3.1. Summary Statistics of Pocket Data for Reference Proteins
3.2. Logistic Regression Model Results
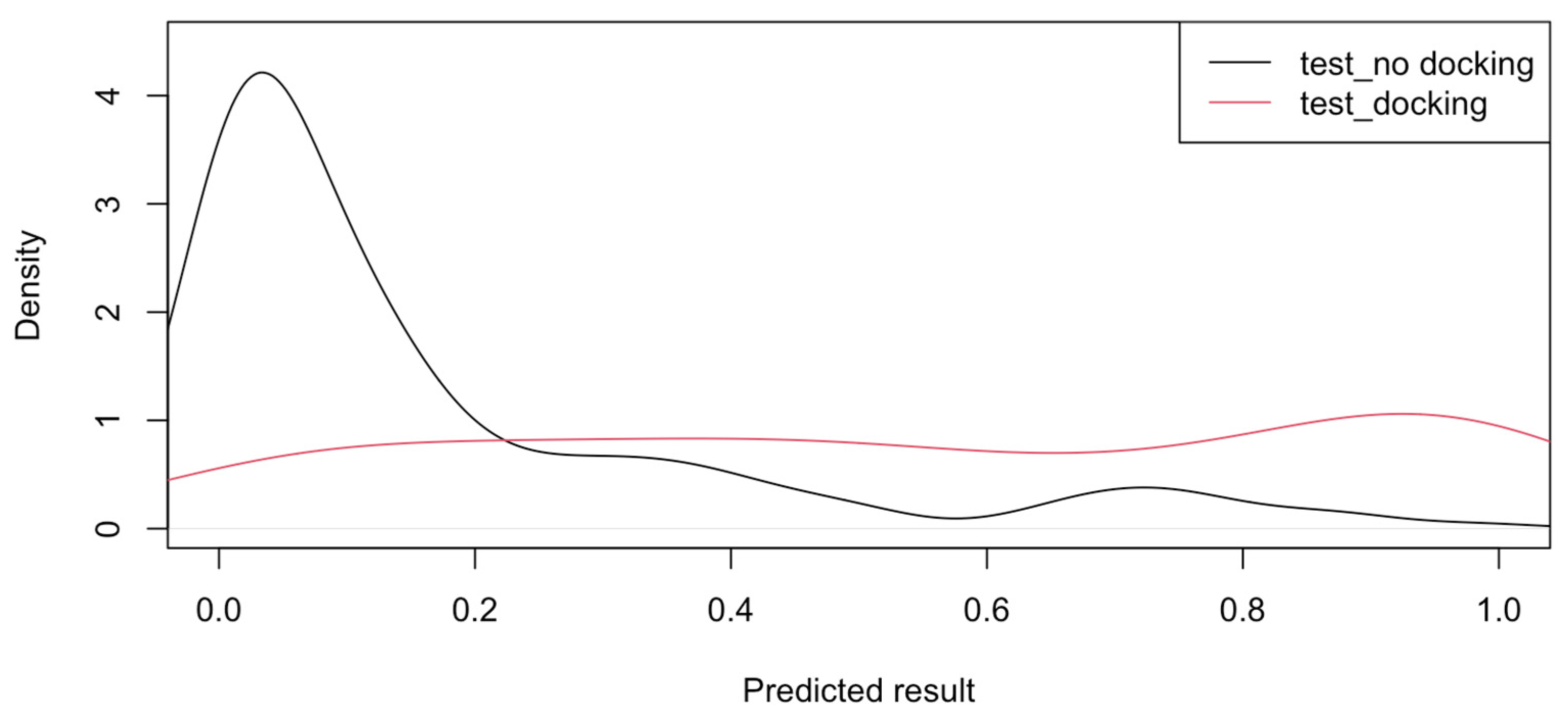
3.3. Predicted Results for Target Proteins
4. Discussion
4.1. Logistic Regression Model
4.2. Druggability Assessment of Target Proteins
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Entry | Protein | Organism | Ligand | Source |
|---|---|---|---|---|
| 1C14 | Ndah | E. coli | Triclosan | [37] |
| 1E9X | Cyp51 | M. tuberculosis | 4-Phenylimidazole | PDB search for similar assemblies |
| 1EA1 | Cyp51 | M. tuberculosis | Fluconazole | [37] |
| 1I2Z | Ndah | E. coli | Imidazole | PDB search for similar assemblies |
| 1KIJ | GyrB | T. thermophilus | Novobiocin | [37] |
| 1KZN | GyrB | E. coli | Clorobiocin | [37] |
| 1QMF | Pbp2X | S. pneumoniae | Carboxylic acid | [37] |
| 1TZ6 | AirK | S. enterica | Aminoimidazole riboside | PDB search for similar assemblies |
| 2Q3J | Cpfc | B. subtilis | N-methyl mesoporphyrin | PDB search for similar assemblies |
| 3DA1 | Gpdh | B. halodurans | Flavin-adenine dinucleotide | [38] |
| 3SYN | FlhF | B. subtilis | Guanosine-5′-diphosphate | PDB search for similar assemblies |
| 3TFC | AroA | L. monocytogenes | Phosphoenolpyruvate | PDB search for Lm protein complexes |
| 3TNL | SkdH | L. monocytogenes | Shikimate and NAD | [38] |
| 3TOZ | SkdH | L. monocytogenes | Nicotinamide-adenine-dinucleotide | PDB search for Lm protein complexes |
| 3U9E | Bup/Acp | L. monocytogenes | Coenzyme A | PDB search for Lm protein complexes |
| 3UF6 | Bup/Acp | L. monocytogenes | 3′-Dephosphocoenzyme A | PDB search for Lm protein complexes |
| 3ZG8 | Pbp4 | L. monocytogenes | Ampicillin | PDB search for Lm protein complexes |
| 3ZG9 | Pbp4 | L. monocytogenes | Cefuroxime | PDB search for Lm protein complexes |
| 3ZGA | Pbp4 | L. monocytogenes | Carbenicillin | PDB search for Lm protein complexes |
| 4INJ | MccF | L. monocytogenes | Methyl sulfamate | PDB search for Lm protein complexes |
| 4JRO | FabG | L. monocytogenes | Nicotinamide-adenine-dinucleotide phosphate | PDB search for Lm protein complexes |
| 4RWW | PstA | L. monocytogenes | Cyclic-di-AMP | PDB search for Lm protein complexes |
| 4S1B | PgpH | L. monocytogenes | Cyclic-di-AMP | PDB search for Lm protein complexes |
| 5B0O | FliH/FliI | S. enterica | Adenosine-5’-diphosphate | PDB search for similar assemblies |
| 5DHP | NadK1 | L. monocytogenes | Novel inhibitor | PDB search for Lm protein complexes |
| 5F1R | PrfA | L. monocytogenes | Ring-fused 2-pyridone (C10) | PDB search for Lm protein complexes |
| 5F7V | Lmo0181 | L. monocytogenes | Cycloalternan | PDB search for Lm protein complexes |
| 5TED | QuiR | L. monocytogenes | Shikimate | PDB search for Lm protein complexes |
| 5UPX | ImpDH | L. monocytogenes | Xanthosine monophosphate | PDB search for Lm protein complexes |
| 5VJD | FbaA | E. coli | Dihydroxyacetonephosphate | [38] |
| 5ZQB | PbpD2 | L. monocytogenes | Penicillin G | PDB search for Lm protein complexes |
| 5ZQC | PbpD2 | L. monocytogenes | Ampicillin | PDB search for Lm protein complexes |
| 5ZQD | PbpD2 | L. monocytogenes | Cefotaxime | PDB search for Lm protein complexes |
| 5ZQE | PbpD2 | L. monocytogenes | Cefuroxime | PDB search for Lm protein complexes |
| 6C5N | IlvC | S. aureus | Hydroxymate inhibitor 1 | [38] |
| 6C8Q | NadE | E. faecalis | Nicotinamide-adenine-dinucleotide | PDB search for similar assemblies |
| 6FXJ | ChdC | L. monocytogenes | Iron coproporphyrin III | PDB search for Lm protein complexes |
| 6HVL | CdaA | L. monocytogenes | Cyclic-di-AMP and AMP | PDB search for Lm protein complexes |
| 6O6N | FasR | M. tuberculosis | Arachinoyl-Coenzyme A | PDB search for similar assemblies |
| 6XXY | LeuB | H. influenzae | O-isobutenyl oxalylhydroxamate | [38] |
| 7NNV | ArgF | M. tuberculosis | Carbamoyl phosphate | PDB search for similar assemblies |
| 7XMD | Cytbo3 | E. coli | Allosteric inhibitor N4 | PDB search for similar assemblies |
| 8EBC | ImpDH | L. monocytogenes | Inosinic acid | PDB search for Lm protein complexes |
| 8H62 | InlA | L. monocytogenes | E-cadherin EC12 | PDB search for Lm protein complexes |
| 8UVZ | DhfR | B. subtilis | Nicotinamide-adenine-dinucleotide and folate | PDB search for similar assemblies |
| 8VDA | FabH | B. subtilis | Coenzyme A | PDB search for similar assemblies |
Appendix B
| PDB Entry | Protein | Function | Source |
|---|---|---|---|
| 1AOD | PlcA | Plays a role in transmembrane signaling. | [35] |
| 1I5N | CheA | A chemotaxis protein involved in the transmission of sensory signals from chemoreceptors to flagellar motors. | [39] |
| 1O6V | InlA | A surface protein that mediates the attachment to and invasion of host cells. | [35] |
| 1XCK | GroeL | Prevents misfolding and promotes refolding and proper assembly of unfolded polypeptides generated under stress conditions. | [40] |
| 1XEU | Inlc | Stimulates the formation of membrane protrusions that mediate the intercellular spread of L. monocytogenes. | [35] |
| 2J70 | RsbU | Acid, antibiotic, cold, ethanol, heat, osmotic and nutritional stress responses require rsbU to activate sigB. | [41] |
| 2PLC | PlcA | Plays a role in transmembrane signaling. | [35] |
| 2WQV | InlB | A surface protein that mediates attachment to and invasion of host cells. | [35] |
| 2ZVY | MotB | A motility protein. | [42] |
| 3B0Z | FlhB | A motility protein; a membrane protein responsible for substrate specificity, switching from rod/hook-type export to filament-type export. | [30] |
| 3FDQ | FlaA | A motility protein; a flagellin protein that polymerizes to form the filaments of bacterial flagella. | [30] |
| 3MIX | FlhA | A motility protein; a membrane protein involved in the flagellar export apparatus. | [30] |
| 4NL2 | Hfq | A regulatory factor involved in the stress response and virulence. | [43] |
| 4UT1 | FlgK | A motility protein; a flagellar hook protein; acts as a hook filament junction protein with flgL to join the flagellar filament to the hook. | [30] |
| 5B0O | FliH/FliI | Involved in type III protein export during flagellum assembly. | [30,44] |
| 5H5T | FliD | A motility protein; a flagellar hook protein; required for morphogenesis and for the elongation of the flagellar filament. | [30] |
| 5LEJ | PrfA | A transcriptional activator of virulence genes. | [35] |
| 5ZIY | FlgL | A motility protein; Lmo0706; acts as a hook filament junction protein with FlgK to join the flagellar filament to the hook. | [30] |
| 6F2D | FliP | A motility protein; forms the core of the central channel in the flagella export apparatus with proteins fliQ and fliR. | [30] |
| 7X1K | DegU | A stress response regulator. | [45] |
| 7X9S | MogR | A transcriptional repressor required for virulence. | [46] |
| 8CQM | PlcB | Plays a role in transmembrane signaling. | [35] |
| 8UMD | FliG | One of three proteins involved in switching the direction of the flagellar rotation. | [30] |
References
- FDA. Common Foodborne Disease Causes. Available online: https://www.fda.gov/files/food/published/Most-Common-Foodborne-Illnesses-%28PDF%29.pdf (accessed on 1 April 2024).
- CDC. National Enteric Disease Surveillance: The Listeria Initiative; National Center for Emerging and Zoonotic Infectious Diseases: Atlanta, GA, USA, 2016; pp. 1–2. [Google Scholar]
- Lotfollahi, L.; Nowrouzi, J.; Irajian, G.; Jazi, F.M.; Kazemi, B.; Eslamian, L.; Falahat, A.; Ramez, M. Prevalence and antimicrobial resistance profiles of Listeria monocytogenes in spontaneous abortions in humans. Afr. J. Microbiol. Res. 2011, 5, 1990–1993. [Google Scholar] [CrossRef]
- CDC Foodborne Germs and Illnesses. Available online: https://www.cdc.gov/food-safety/about/index.html (accessed on 27 March 2024).
- Bucur, F.I.; Grigore-Gurgu, L.; Crauwels, P.; Riedel, C.U.; Nicolau, A.I. Resistance of Listeria monocytogenes to stress conditions encountered in food and food processing environments. Front. Microbiol. 2018, 9, 2700. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, A.; Fu, M.; Wang, A.; Chen, K.; Jia, Q.; Huang, Z. Investigation of incidents and trends of antimicrobial resistance in foodborne pathogens in eight countries from historical sample data. Int. J. Environ. Res. Public Health 2020, 17, 472. [Google Scholar] [CrossRef] [PubMed]
- Abd Al-Mayahi, F.S.; Jaber, S.M. Multiple drug resistance of Listeria monocytogenes isolated from aborted women by using serological and molecular techniques in Diwaniyah city/Iraq. Iran. J. Microbiol. 2020, 12, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Rantsiou, K.; Mataragas, M.; Alessandria, V.; Cocolin, L. Expression of virulence genes of Listeria monocytogenes in food. J. Food Saf. 2012, 32, 161–168. [Google Scholar] [CrossRef]
- Charpentier, E.; Courvalin, P. Antibiotic resistance in Listeria spp. Antimicrob. Agents Chemother. 1999, 43, 2103–2108. [Google Scholar] [CrossRef]
- Hanes, R.M.; Huang, Z. Investigation of Antimicrobial Resistance Genes in Listeria monocytogenes from 2010 through to 2021. Int. J. Environ. Res. Public Health 2022, 19, 5506. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef]
- Zhang, F.; Graham, J.; Zhai, T.; Liu, Y.; Huang, Z. Discovery of MurA inhibitors as novel antimicrobials through an integrated computational and experimental approach. Antibiotics 2022, 11, 528. [Google Scholar] [CrossRef]
- Hecker, M.; Pané-Farré, J.; Volker, U. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu. Rev. Microbiol. 2007, 61, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W. Bacterial Pathogenesis. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. Available online: https://www.ncbi.nlm.nih.gov/books/NBK8526/ (accessed on 13 March 2023).
- CDC. Antibiotic Resistance Threats in the United States; U.S Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Cui, K.; Gong, I.; Dong, A.; Yan, J.; Wang, M.; Huang, Z. Investigation of virulence genes detected in antimicrobial-resistance pathogens isolates for five countries across the world. Processes 2020, 8, 1589. [Google Scholar] [CrossRef]
- Gashaw, I.; Ellinghaus, P.; Sommer, A.; Asadullah, K. What makes a good drug target? Drug Discov. Today 2011, 16, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Kufareva, I. The flexible pocketome engine for structural chemogenomics. Methods Mol. Biol. 2009, 575, 249–279. [Google Scholar] [PubMed]
- Zhang, F.; Zhai, T.; Haider, S.; Liu, Y.; Huang, Z. Synergistic effect of chlorogenic acid and caffeic acid with fosfomycin in growth inhibition of a resistant Listeria monocytogenes strain. ACS Omega 2020, 5, 7537–7544. [Google Scholar] [CrossRef]
- Sheridan, R.P.; Maiorov, V.N.; Holloway, M.K.; Cornell, W.D.; Gao, Y. Drug-like density: A method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the Protein Data Bank. J. Chem. Inf. Model. 2010, 50, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Maxim, T.; Abagyan, R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell Proteom. 2005, 4, 752–761. [Google Scholar] [CrossRef]
- Molsoft MolScreen. Available online: https://www.molsoft.com/molscreen.html (accessed on 1 April 2024).
- Molsoft ICM-Pro. Available online: https://www.molsoft.com/icm_pro.html (accessed on 1 April 2024).
- Molsoft ICM User’s Guide. Available online: https://www.molsoft.com/gui/3d-predict.html#3d-predict-tools-identify-ligand-binding-pocket (accessed on 1 April 2024).
- RSCB PDB. Available online: https://www.rcsb.org/pages/about-us/index (accessed on 1 April 2024).
- PDB-101. Available online: https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/introduction (accessed on 1 April 2024).
- Moorhead, S.M.; Dykes, G.A. The role of the sigB gene in the general stress response of Listeria monocytogenes varies between a strain of serotype 1/2a and a strain of serotype 4c. Curr. Microbiol. 2003, 46, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Good, J.A.D.; Andersson, C.; Hansen, S.; Wall, J.; Krishnan, K.S.; Begum, A.; Grundström, C.; Niemiec, M.S.; Vaitkevicius, K.; Chorell, E.; et al. Attenuating Listeria monocytogenes virulence by targeting the regulatory protein PrfA. Cell Chem. Biol. 2016, 23, 404–414. [Google Scholar] [CrossRef]
- Hanes, R.; Fangyuan, Z.; Huang, Z. Protein interaction network analysis to investigate stress response, virulence, and antibiotic resistance mechanisms in Listeria monocytogenes. Microorganisms 2023, 11, 930. [Google Scholar] [CrossRef]
- Stogios, P.J. R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 20 February 2022).
- Sarkar, S.K.; Midi, H. Multicollinearity problems and remedies in binary logistic regression. Res. Bull. Inst. Math. Res. 2010, 3, 27–34. [Google Scholar]
- Sperandei, S. Understanding logistic regression analysis. Biochem. Med. 2014, 24, 12–18. [Google Scholar] [CrossRef]
- Hughes, G.; Choudhury, R.A.; McRoberts, N. Summary measures of predictive power associated with logistic regression models of disease risk. Phytopathology 2019, 109, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Matle, I.; Khanyisile, R.M.; Evelyn, M. A review of Listeria monocytogenes from meat and meat products: Epidemiology, virulence factors, antimicrobial resistance, and diagnosis. Onderstepoort J. Vet. Res. 2020, 87, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. The role of binding kinetics in therapeutically useful drug action. Curr. Opin. Drug Discov. Dev. 2009, 12, 31–39. [Google Scholar] [CrossRef]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef]
- Palumbo, M.; Sosa, E.J.; Castello, F.; Schottlender, G.; Serral, F.; Turjanski, A.G.; Palomino, M. Integrating diverse layers of omic data to identify novel drug targets in Listeria monocytogenes. Front. Drug Discov. 2022, 2, 969415. [Google Scholar] [CrossRef]
- Dons, L.; Eriksson, E.; Jin, Y.; Rottenberg, M.E.; Kristensson, K.; Larsen, C.N.; Bresciani, J.; Olsen, J.E. Role of flagellin and the two-component CheA/CheY system of Listeria monocytogenes in host cell invasion and virulence. Infect. Immun. 2004, 72, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Fourie, K.R.; Wilson, H.L. Understanding GroEL and DnaK Stress Response Proteins as Antigens for Bacterial Diseases. Vaccines 2020, 8, 773. [Google Scholar] [CrossRef]
- Shin, J.; Brody, M.S.; Price, C.W. Physical and antibiotic stresses require activation of the RsbU phosphatase to induce the general stress response in Listeria monocytogenes. Microbiology 2010, 156, 2660–2669. [Google Scholar] [CrossRef]
- Hingston, P.A.; Piercey, M.J.; Truelstrup Hansen, L. Genes associated with desiccation and osmotic stress in Listeria monocytogenes as revealed by insertional mutagenesis. Appl. Environ. Microbiol. 2015, 81, 5350–5362. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.K.; Larsen, M.H.; Ingmer, H.; Søgaard-Andersen, L.; Kallipolitis, B.H. The RNA-binding protein Hfq of Listeria monocytogenes: Role in stress tolerance and virulence. J. Bacteriol. 2004, 186, 3355–3362. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Pagniez, H.; Botton, E.; Fréhel, C.; Dubail, I.; Jacquet, C.; Charbit, A.; Raynaud, C. Role of FliF and FliI of Listeria monocytogenes in flagellar assembly and pathogenicity. Infect. Immun. 2005, 73, 5530–5539. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.M.; Olsen, J.E.; Dons, L. Characterization of DegU, a response regulator in Listeria monocytogenes, involved in regulation of motility and contributes to virulence. FEMS Microbiol Lett. 2004, 240, 171–179. [Google Scholar] [CrossRef]
- Gründling, A.; Burrack, L.S.; Archie Bouwer, H.G.; Higgins, D.E. Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc. Natl. Acad. Sci. USA 2004, 101, 12318–12323. [Google Scholar] [CrossRef]

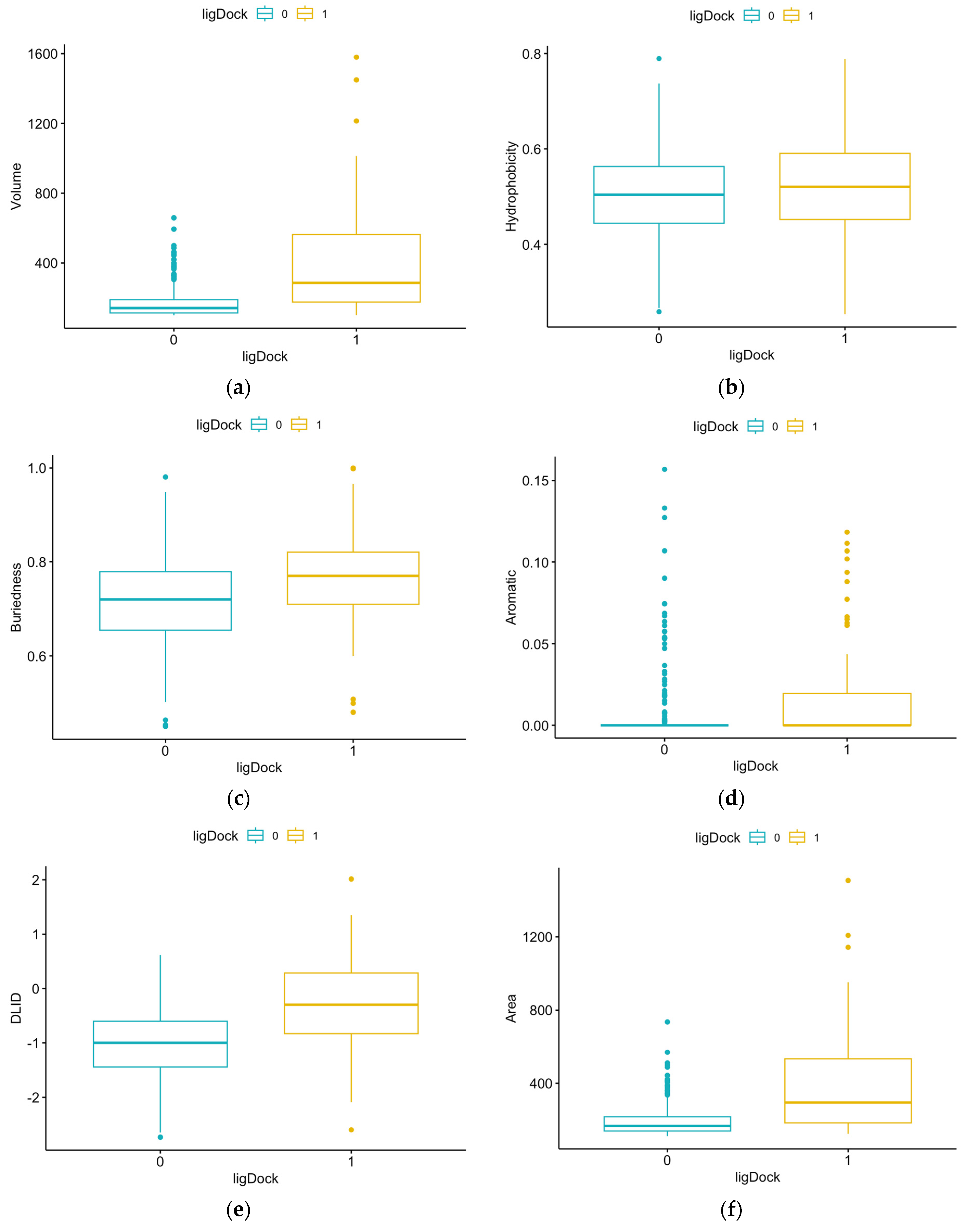
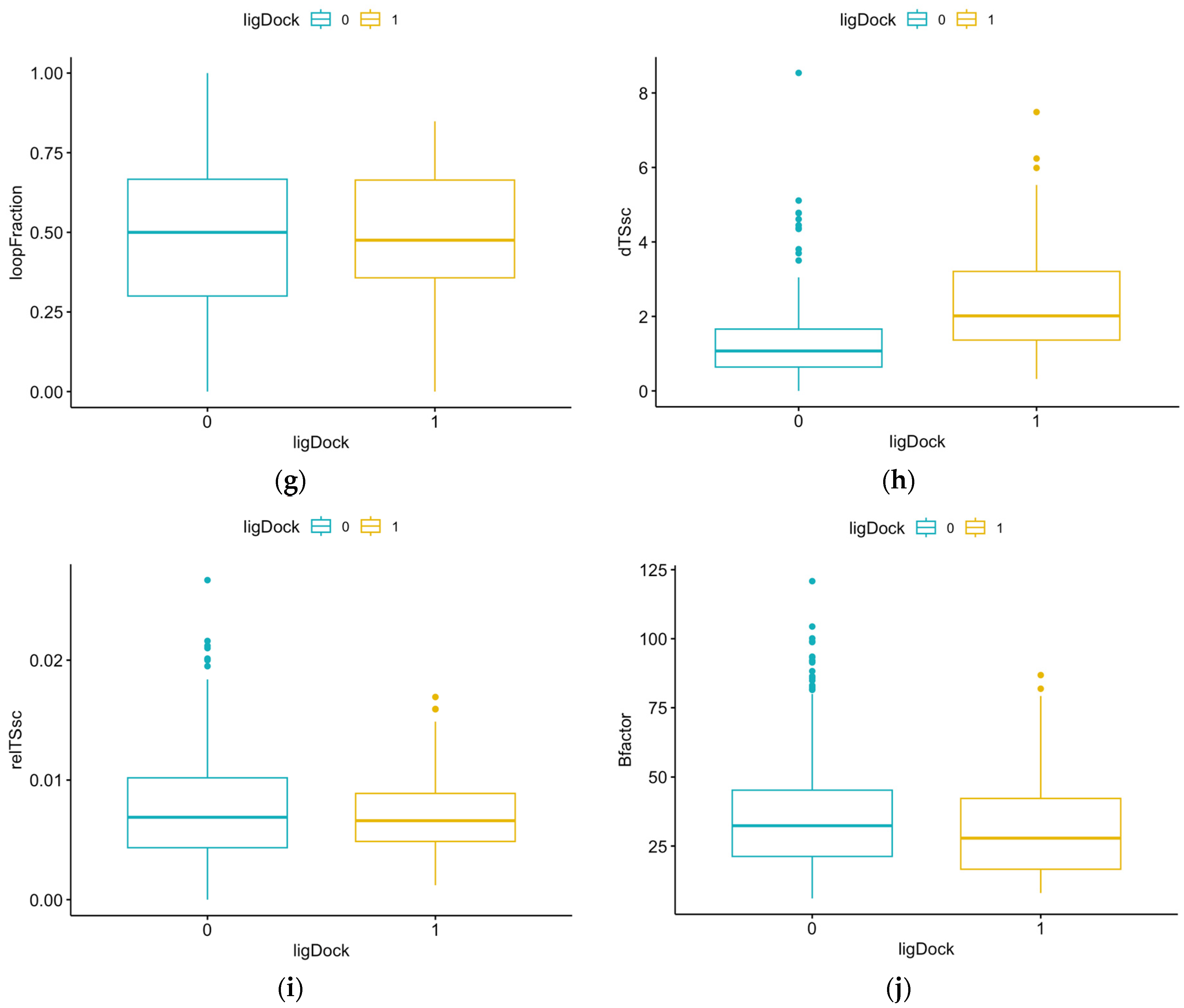
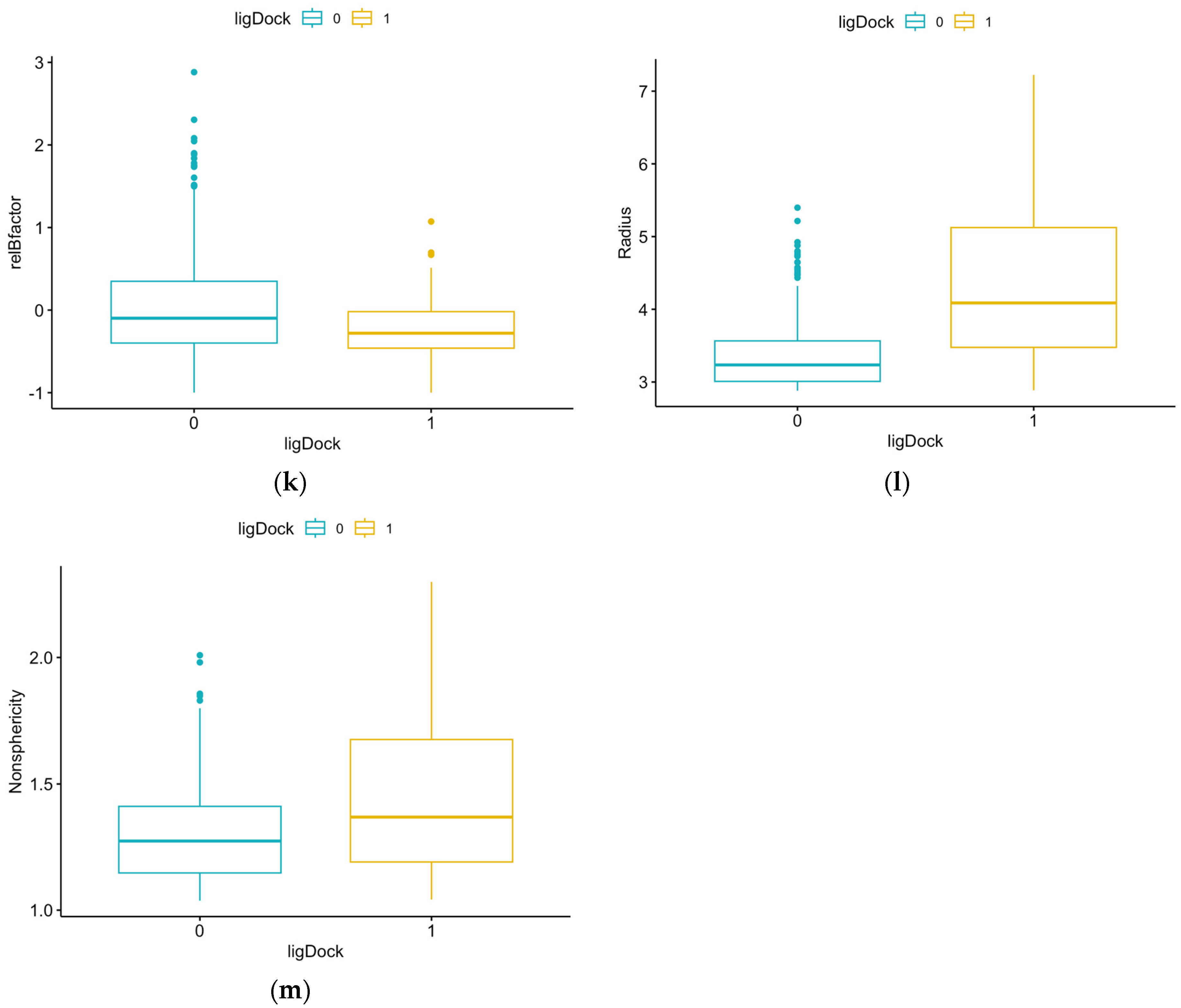

| Parameter | Definition |
|---|---|
| Volume | Pocket volume |
| Hydrophobicity | Percentage of pocket surface in contact with hydrophobic protein residues (in the range of 0–1) |
| Buriedness | Ratio of pocket surface area covered by its shell to total pocket surface area (1.0 means completely buried) |
| Aromatic | Fraction of pocket formed by aromatic side chains (higher is better) |
| DLID | Drug-like density score measuring bindability of proteins (slightly negative and above 0 are considered “druggable”) |
| Area | Pocket area |
| LoopFraction | Fraction of pocket formed by residues from loops (lower is better) |
| dTSsc | Estimate of entropic penalty associated with flexible side chains forming parts of the pocket |
| relTSsc | Same as dTSsc but relative to pocket volume (lower is better) |
| Bfactor | Average b-factor of pocket-forming atoms (lower is better) |
| relBfactor | Normalized deviation of pocket b-factor from the average over the protein (lower is better) |
| Radius | Radius of an ideal spherical cavity with the same volume as the pocket |
| Nonsphericity | Ratio of pocket area to ideal spherical cavity area (1.0 means completely spherical) |
| Variable (xi) | Coefficient (βi) | Std. Error | z Value | Pr (>|z|) |
|---|---|---|---|---|
| (Intercept) | 3.9724 | 1.8218 | 2.180 | 0.029277 |
| Hydrophobicity | −15.0826 | 4.2276 | −3.568 | 0.000360 |
| Aromatic | −1.6691 | 1.8166 | −0.919 | 0.358196 |
| Buriedness | −14.8371 | 5.5163 | −2.690 | 0.007152 |
| DLID | 31.8352 | 10.1678 | 3.131 | 0.001742 |
| Area | 15.0366 | 12.9474 | 1.161 | 0.245494 |
| loopFraction | −1.8166 | 1.1914 | −1.525 | 0.127343 |
| dTSsc | −7.0218 | 5.4238 | −1.295 | 0.195450 |
| relTSsc | 5.9622 | 3.8447 | 1.551 | 0.120960 |
| Bfactor | 0.6733 | 3.4303 | 0.196 | 0.844396 |
| relBfactor | −10.5689 | 3.0309 | −3.487 | 0.000488 |
| Nonsphericity | −9.0507 | 4.0022 | −2.261 | 0.023732 |
| Cutoff Value | Accuracy 1 | Sensitivity 2 | Specificity 3 | Positive Pred. Rate 4 | Negative Pred. Rate 5 |
|---|---|---|---|---|---|
| 0.60 | 0.7790 | 0.8047 | 0.6731 | 0.9105 | 0.4545 |
| 0.50 | 0.8015 | 0.8309 | 0.7000 | 0.9503 | 0.5455 |
| 0.40 | 0.8015 | 0.8586 | 0.6579 | 0.8632 | 0.6494 |
| 0.30 | 0.7715 | 0.8644 | 0.5889 | 0.8053 | 0.6883 |
| 0.20 | 0.7491 | 0.9020 | 0.5439 | 0.7263 | 0.8052 |
| Prediction | Reference | |
|---|---|---|
| 0 | 1 | |
| 0 | 172 | 18 |
| 1 | 35 | 42 |
| ligDock = 0 | ligDock = 1 (Druggable) |
|---|---|
| 1I5N (CheA) 1XEU (InlC) 2J70 (RsBU) 2PLC (PlcA) 2WQV (InlB) 2ZVY (MotB) 3FDQ (FlaA) 3MIX (FlhA) 4NL2 (Hfq) 5H5T (FliD) 6F2D (Flip) 7X1K (DegU) 8CQM (PlcB) | 1O6V (InlA) 1XCK (GroEL) 3B0Z (FlhB) 4UT1 (FlgK) 5B0O (FliH/FliI complex) 5LEJ (PrfA) 5ZIY (FlgL) 7X9S (MogR) 8UMD (FliG) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanes, R.; Liu, Y.; Huang, Z. Druggability Analysis of Protein Targets for Drug Discovery to Combat Listeria monocytogenes. Microorganisms 2024, 12, 1073. https://doi.org/10.3390/microorganisms12061073
Hanes R, Liu Y, Huang Z. Druggability Analysis of Protein Targets for Drug Discovery to Combat Listeria monocytogenes. Microorganisms. 2024; 12(6):1073. https://doi.org/10.3390/microorganisms12061073
Chicago/Turabian StyleHanes, Robert, Yanhong Liu, and Zuyi Huang. 2024. "Druggability Analysis of Protein Targets for Drug Discovery to Combat Listeria monocytogenes" Microorganisms 12, no. 6: 1073. https://doi.org/10.3390/microorganisms12061073