
Assessing Antibiotic-Resistant Genes in University Dormitory Washing Machines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Metagenomic Assembly, ARG and MGE Analysis
2.4. Statistical Analysis and Result Visualization
3. Results and Discussion
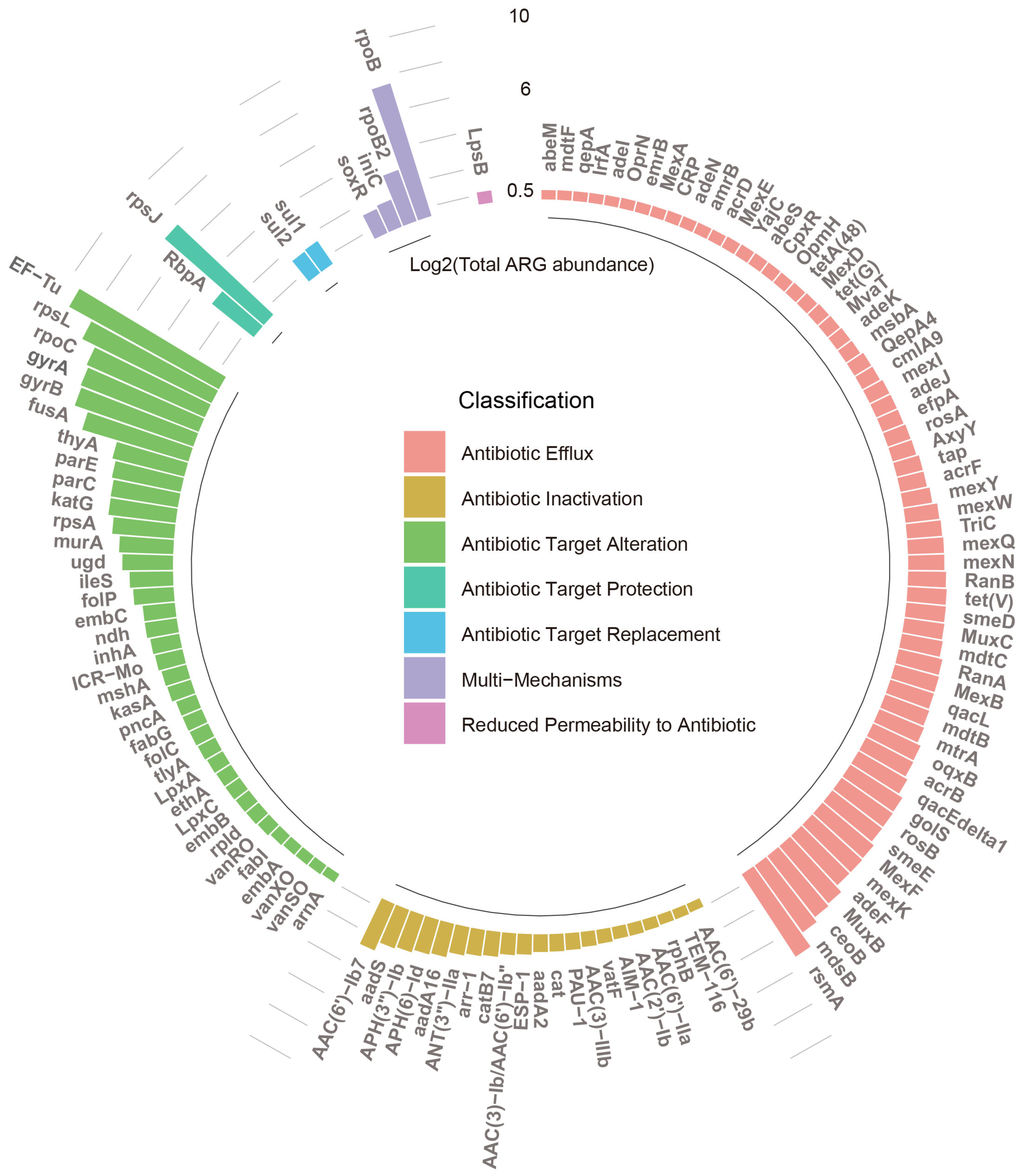
3.1. Diversity and Abundance of ARGs
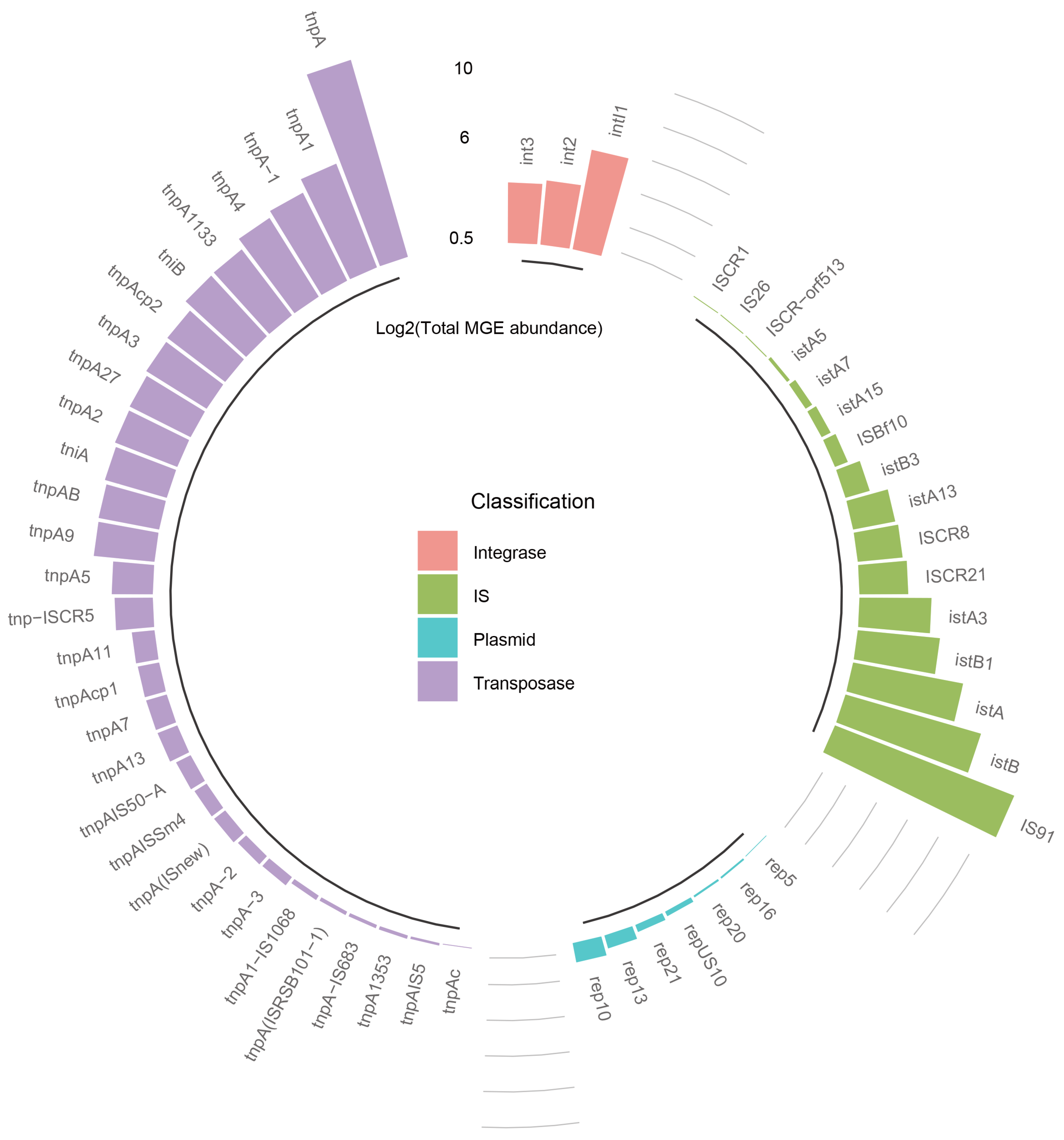
3.2. Diversity and Abundance of MGEs
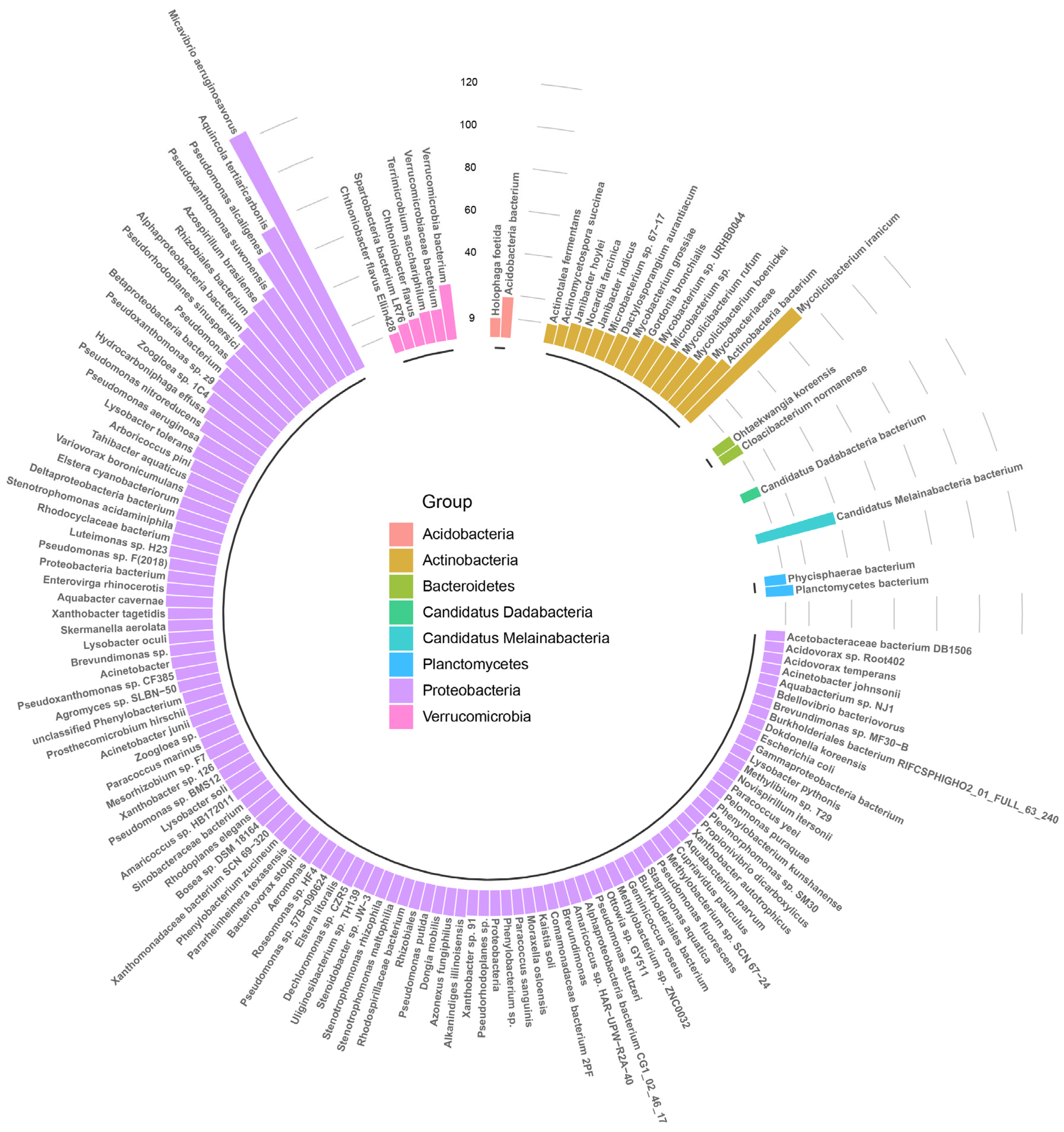
3.3. Characterization of Bacterial Community
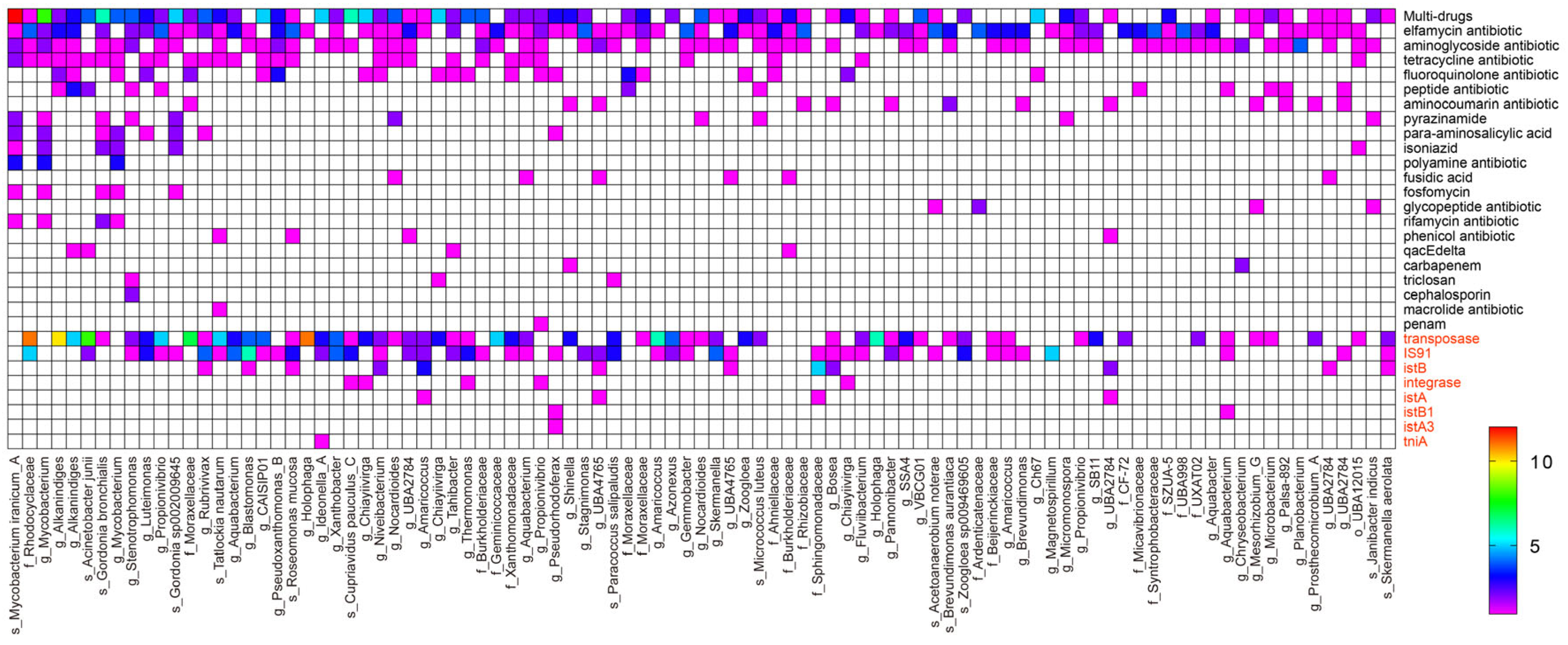
3.4. The Abundance and Occurrence of Microbial Communities Associated with the ARGs and MGEs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zinn, M.-K.; Flemming, H.-C.; Bockmühl, D. A comprehensive view of microbial communities in the laundering cycle suggests a preventive effect of soil bacteria on malodour formation. Microorganisms 2022, 10, 1465. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, S.; Yang, J.; Li, Y.; Kogure, E.; Zhu, Y.; Xiong, W.; Chen, E.; Shi, G. Metabarcoding Analysis of Microorganisms Inside Household Washing Machines in Shanghai, China. Microorganisms 2024, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Jacksch, S.; Zohra, H.; Weide, M.; Schnell, S.; Egert, M. Cultivation-Based Quantification and Identification of Bacteria at Two Hygienic Key Sides of Domestic Washing Machines. Microorganisms 2021, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Bockmuehl, D.P.; Schages, J.; Rehberg, L. Laundry and textile hygiene in healthcare and beyond. Microb. Cell 2019, 6, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Callewaert, C.; Van Nevel, S.; Kerckhof, F.-M.; Granitsiotis, M.S.; Boon, N. Bacterial Exchange in Household Washing Machines. Front. Microbiol. 2015, 6, 1381. [Google Scholar] [CrossRef] [PubMed]
- Jacksch, S.; Kaiser, D.; Weis, S.; Weide, M.; Ratering, S.; Schnell, S.; Egert, M. Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines. Microorganisms 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Schmithausen, R.M.; Sib, E.; Exner, M.; Hack, S.; Rösing, C.; Ciorba, P.; Bierbaum, G.; Savin, M.; Bloomfield, S.F.; Kaase, M.; et al. The Washing Machine as a Reservoir for Transmission of Extended-Spectrum-Beta-Lactamase (CTX-M-15)-Producing Klebsiella oxytoca ST201 to Newborns. Appl. Environ. Microbiol. 2019, 85, e01435-19. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef]
- von Wintersdorff, C.J.H.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, B.; Yu, G. Antibiotic resistance genes in China: Occurrence, risk, and correlation among different parameters. Environ. Sci. Pollut. Res. 2018, 25, 21467–21482. [Google Scholar] [CrossRef]
- Schages, L.; Lucassen, R.; Wichern, F.; Kalscheuer, R.; Bockmuehl, D. The Household Resistome: Frequency of beta-Lactamases, Class 1 Integrons, and Antibiotic-Resistant Bacteria in the Domestic Environment and Their Reduction during Automated Dishwashing and Laundering. Appl. Environ. Microbiol. 2020, 86, e02062-20. [Google Scholar] [CrossRef]
- Rehberg, L.; Frontzek, A.; Melhus, A.; Bockmuehl, D.P. Prevalence of beta-lactamase genes in domestic washing machines and dishwashers and the impact of laundering processes on antibiotic-resistant bacteria. J. Appl. Microbiol. 2017, 123, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, L.C.; Chai, Y.H.; Xu, J.F. Advantages and challenges of metagenomic sequencing for the diagnosis of pulmonary infectious diseases. Clin. Respir. J. 2022, 16, 646–656. [Google Scholar] [CrossRef]
- Dai, D.; Brown, C.; Bürgmann, H.; Larsson, D.G.J.; Nambi, I.; Zhang, T.; Flach, C.-F.; Pruden, A.; Vikesland, P.J. Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 2022, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Begmatov, S.; Beletsky, A.V.; Dorofeev, A.G.; Pimenov, N.V.; Mardanov, A.V.; Ravin, N.V. Metagenomic insights into the wastewater resistome before and after purification at large-scale wastewater treatment plants in the Moscow city. Sci. Rep. 2024, 14, 6349. [Google Scholar] [CrossRef]
- Qiu, T.; Huo, L.; Guo, Y.; Gao, M.; Wang, G.; Hu, D.; Li, C.; Wang, Z.; Liu, G.; Wang, X. Metagenomic assembly reveals hosts and mobility of common antibiotic resistome in animal manure and commercial compost. Environ. Microbiome 2022, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Munk, P.; Brinch, C.; Møller, F.D.; Petersen, T.N.; Hendriksen, R.S.; Seyfarth, A.M.; Kjeldgaard, J.S.; Svendsen, C.A.; van Bunnik, B.; Berglund, F.; et al. Genomic analysis of sewage from 101 countries reveals global landscape of antimicrobial resistance. Nat. Commun. 2022, 13, 7251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 15175. [Google Scholar] [CrossRef]
- Zhou, G.; Tao, H.-B.; Wen, X.; Wang, Y.-S.; Peng, H.; Liu, H.-Z.; Yang, X.-J.; Huang, X.-M.; Shi, Q.-S.; Xie, X.-B. Metagenomic analysis of microbial communities and antibiotic resistance genes in spoiled household chemicals. Chemosphere 2022, 291, 132766. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Pärnänen, K.; Karkman, A.; Hultman, J.; Lyra, C.; Bengtsson-Palme, J.; Larsson, D.J.; Rautava, S.; Isolauri, E.; Salminen, S.; Kumar, H. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018, 9, 3891. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.A.; Michelitsch, L.-M.; Glawe, A.J.; Lee, H.; Huttelmaier, S.; Hellgeth, N.; Ben Maamar, S.; Hartmann, E.M. Toothbrush microbiomes feature a meeting ground for human oral and environmental microbiota. Microbiome 2021, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, X.; Shi, Z.-Y.; Fan, X.-Y.; Zhou, Z.-W. Bacterial communities, potential pathogens and antibiotic resistance genes of silver-loaded stainless steel pipe wall biofilm in domestic hot water system. J. Water Process. Eng. 2021, 40, 101935. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, B.; Mo, X.; Li, P.; Li, B.; Li, Q.; Li, N.; Mo, S.; Ou, Q.; Shen, P. Prevalence and proliferation of antibiotic resistance genes in the subtropical mangrove wetland ecosystem of South China Sea. MicrobiologyOpen 2019, 8, e871. [Google Scholar] [CrossRef]
- Pourahmad Jaktaji, R.; Lesani, S.M.; Akhavan, H.; Tanhaei, M. The Role of Some Transcription Factors in Expression of GyrA and GyrB Following Exposure to Ciprofloxacin. Jundishapur J. Microbiol. 2020, 13, e100654. [Google Scholar] [CrossRef]
- Knopp, M.; Andersson, D.I. Predictable Phenotypes of Antibiotic Resistance Mutations. mBio 2018, 9, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Andre, E.; Goeminne, L.; Cabibbe, A.; Beckert, P.; Kabamba Mukadi, B.; Mathys, V.; Gagneux, S.; Niemann, S.; Van Ingen, J.; Cambau, E. Consensus numbering system for the rifampicin resistance-associated rpoB gene mutations in pathogenic mycobacteria. Clin. Microbiol. Infect. 2017, 23, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.-N.; Chen, H.; Gao, R.-X.; Zhu, Y.-G.; Rensing, C. Organic compounds stimulate horizontal transfer of antibiotic resistance genes in mixed wastewater treatment systems. Chemosphere 2017, 184, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-C.; Gao, J.-F.; Zhang, S.-J.; Gao, Y.-Q.; Sun, L.-X. Emergence and spread patterns of antibiotic resistance genes during two different aerobic granular sludge cultivation processes. Environ. Int. 2020, 137, 105540. [Google Scholar] [CrossRef]
- Guo, J.; Li, J.; Chen, H.; Bond, P.L.; Yuan, Z. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Shi, B.; Bai, Y.; Wang, D. Bacterial community of biofilms developed under different water supply conditions in a distribution system. Sci. Total Environ. 2014, 472, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Baca, M.; Piekarska, K. Biodiversity of organisms inhabiting the water supply network of Wroclaw. Detection of pathogenic organisms constituting a threat for drinking water recipients. Sci. Total Environ. 2020, 715, 136732. [Google Scholar] [CrossRef]
- Gu, X.; Zhai, H.; Cheng, S. Fate of antibiotics and antibiotic resistance genes in home water purification systems. Water Res. 2021, 190, 116762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, W.; Chen, J.; Wang, F.; Qi, W.; Li, Y.; Xie, B. Effect of hydraulic conditions on the prevalence of antibiotic resistance in water supply systems. Chemosphere 2019, 235, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. Biomed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.S.; Sahu, M.K.; Kandasamy, K. Isolation and characterisation of streptomycetes producing antibiotic, from a mangrove environment. Asian J. Microbiol. Biotechnol. Environ. Sci. 2005, 7, 457–464. [Google Scholar] [CrossRef]
- Sénéchal, A.; Valour, F.; Chidiac, C.; Ferry, T. Pulmonary actinomycosis with large cavitation in an alcoholic 39-year-old man. BMJ Case Rep. 2014, 2014, bcr2014206556. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Yang, J.; Kim, S.; Park, J.; Kim, M.; Park, W. Occurrence of antibiotic resistance genes and multidrug-resistant bacteria during wastewater treatment processes. Sci. Total Environ. 2022, 811, 152331. [Google Scholar] [CrossRef]
- Boxberger, M.; Ben Khedher, M.; Magnien, S.; Cassir, N.; La Scola, B. Draft genome and description of Chryseobacterium manosquense strain Marseille-Q2069T sp. nov., a new bacterium isolated from human healthy skin. New Microbes New Infect. 2020, 38, 100805. [Google Scholar] [CrossRef]
- Nicholson, A.C.; Gulvik, C.A.; Whitney, A.M.; Humrighouse, B.W.; Bell, M.E.; Holmes, B.; Steigerwalt, A.G.; Villarma, A.; Sheth, M.; Batra, D.; et al. Division of the genus Chryseobacterium: Observation of discontinuities in amino acid identity values, a possible consequence of major extinction events, guides transfer of nine species to the genus Epilithonimonas, eleven species to the genus Kaistella, and three species to the genus Halpernia gen. nov., with description of Kaistella daneshvariae sp. nov. and Epilithonimonas vandammei sp. nov. derived from clinical specimens. Int. J. Syst. Evol. Microbiol. 2020, 70, 4432–4450. [Google Scholar] [CrossRef] [PubMed]
- Vaneechoutte, M.; Kaempfer, P.; De Baere, T.; Avesani, V.; Janssens, M.; Wauters, G. Chryseobacterium hominis sp nov., to accommodate clinical isolates biochemically similar to CDC groups II-h and II-c. Int. J. Syst. Evol. Microbiol. 2007, 57, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Balada-Llasat, J.-M.; Elkins, C.; Swyers, L.; Bannerman, T.; Pancholi, P. Pseudo-Outbreak of Cupriavidus pauculus Infection at an Outpatient Clinic Related to Rinsing Culturette Swabs in Tap Water. J. Clin. Microbiol. 2010, 48, 2645–2647. [Google Scholar] [CrossRef] [PubMed]
- Huda, S.A.; Yadava, S.; Kahlown, S.; Jilani, M.H.; Sharma, B. A Rare Case of Ventilator-Associated Pneumonia Caused by Cupriavidus pauculus. Cureus 2020, 12, e8573. [Google Scholar] [CrossRef] [PubMed]
- García-Masedo Fernández, S.; García, J.; Sáez-Nieto, J.A.; Orden, B. Bacteriemia nosocomial por Cupriavidus pauculus en paciente con colitis ulcerosa. Enfermedades Infecc. Y Microbiol. Clínica 2019, 37, 677–678. [Google Scholar] [CrossRef]
- Aydin, B.; Dilli, D.; Zenciroglu, A.; Okumus, N.; Ozkan, S.; Tanir, G. A case of newborn with community acquired pneumonia caused by Cupriavidus pauculus. Tuberk. Ve Toraks 2012, 60, 160–162. [Google Scholar] [CrossRef]
- Tian, S.; Zhu, B.; Tian, Y.; Li, J.; Peng, C. Histiocytic Necrotizing Lymphadenitis with Cupriavidus Pauculus Infection in a Patient with Graves Hyperthyroidism: A Case Report. Infect. Drug Resist. 2022, 15, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Kadouri, D.; Venzon, N.C.; O’Toole, G.A. Vulnerability of pathogenic biofilms to Micavibrio aeruginosavorus. Appl. Environ. Microbiol. 2007, 73, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kadouri, D.E.; Wu, M. Genomic insights into an obligate epibiotic bacterial predator: Micavibrio aeruginosavorus ARL-13. BMC Genom. 2011, 12, 453. [Google Scholar] [CrossRef]
- Romanowski, E.G.; Gupta, S.; Pericleous, A.; Kadouri, D.E.; Shanks, R.M.Q. Clearance of Gram-Negative Bacterial Pathogens from the Ocular Surface by Predatory Bacteria. Antibiotics 2021, 10, 810. [Google Scholar] [CrossRef]
- Shanks, R.M.Q.; Davra, V.R.; Romanowski, E.G.; Brothers, K.M.; Stella, N.A.; Godboley, D.; Kadouri, D.E. An Eye to a Kill: Using Predatory Bacteria to Control Gram-Negative Pathogens Associated with Ocular Infections. PLoS ONE 2013, 8, e66723. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lemenze, A.; Donnelly, R.J.; Connell, N.D.; Kadouri, D.E. Keeping it together: Absence of genetic variation and DNA incorporation by the predatory bacteria Micavibrio aeruginosavorus and Bdellovibrio bacteriovorus during predation. Res. Microbiol. 2018, 169, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, H.; Daley, C.; Gitti, Z.; Hashemi, A.; Heidarieh, P.; Moore, E.R.B.; Naser, A.D.; Russo, C.; van Ingen, J.; Tortoli, E. Mycobacterium iranicum sp nov., a rapidly growing scotochromogenic species isolated from clinical specimens on three different continents. Int. J. Syst. Evol. Microbiol. 2013, 63, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Mizutani, M.; Nagahara, Y.; Asano, M.; Masamoto, D.; Sawada, O.; Aono, A.; Chikamatsu, K.; Mitarai, S. Successful Treatment of Peritoneal Dialysis-related Peritonitis due to Mycobacterium iranicum. Intern. Med. 2016, 55, 1929–1931. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Singh, S. Non-tuberculous mycobacteria in TB-endemic countries: Are we neglecting the danger? PLoS Negl. Trop. Dis. 2010, 4, e615. [Google Scholar] [CrossRef] [PubMed]
- Lechner, U.; Brodkorb, D.; Geyer, R.; Hause, G.; Hartig, C.; Auling, G.; Fayolle-Guichard, F.; Piveteau, P.; Muller, R.H.; Rohwerder, T. Aquincola tertiaricarbonis gen. nov., sp nov., a tertiary butyl moiety-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2007, 57, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.H.; Rohwerder, T.; Harms, H. Degradation of fuel oxygenates and their main intermediates by Aquincola tertiaricarbonis L108. Microbiology 2008, 154, 1414–1421. [Google Scholar] [CrossRef]
- Pourmazaheri, H.; Jouzani, G.S.; Karimi, E.; Nekouei, S.M.K.; Tabatabaei, M.; Amiri, R.M. Development of a bioprocess for fast production of enriched biocompost from municipal solid wastes. Int. Biodeterior. Biodegrad. 2015, 104, 482–489. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Zhang, Y.; Mi, J. Assessing Antibiotic-Resistant Genes in University Dormitory Washing Machines. Microorganisms 2024, 12, 1112. https://doi.org/10.3390/microorganisms12061112
Chen W, Zhang Y, Mi J. Assessing Antibiotic-Resistant Genes in University Dormitory Washing Machines. Microorganisms. 2024; 12(6):1112. https://doi.org/10.3390/microorganisms12061112
Chicago/Turabian StyleChen, Wenbo, Yu Zhang, and Jiandui Mi. 2024. "Assessing Antibiotic-Resistant Genes in University Dormitory Washing Machines" Microorganisms 12, no. 6: 1112. https://doi.org/10.3390/microorganisms12061112
APA StyleChen, W., Zhang, Y., & Mi, J. (2024). Assessing Antibiotic-Resistant Genes in University Dormitory Washing Machines. Microorganisms, 12(6), 1112. https://doi.org/10.3390/microorganisms12061112