
A Novel View on the Taxonomy of Sulfate-Reducing Bacterium ‘Desulfotomaculum salinum’ and a Description of a New Species Desulfofundulus salinus sp. nov.

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of Bacterial Strains
2.2. Media Composition and Isolation
2.3. Phenotypic Characterization
2.4. The 16S rRNA Gene and Genome Sequencing and Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results
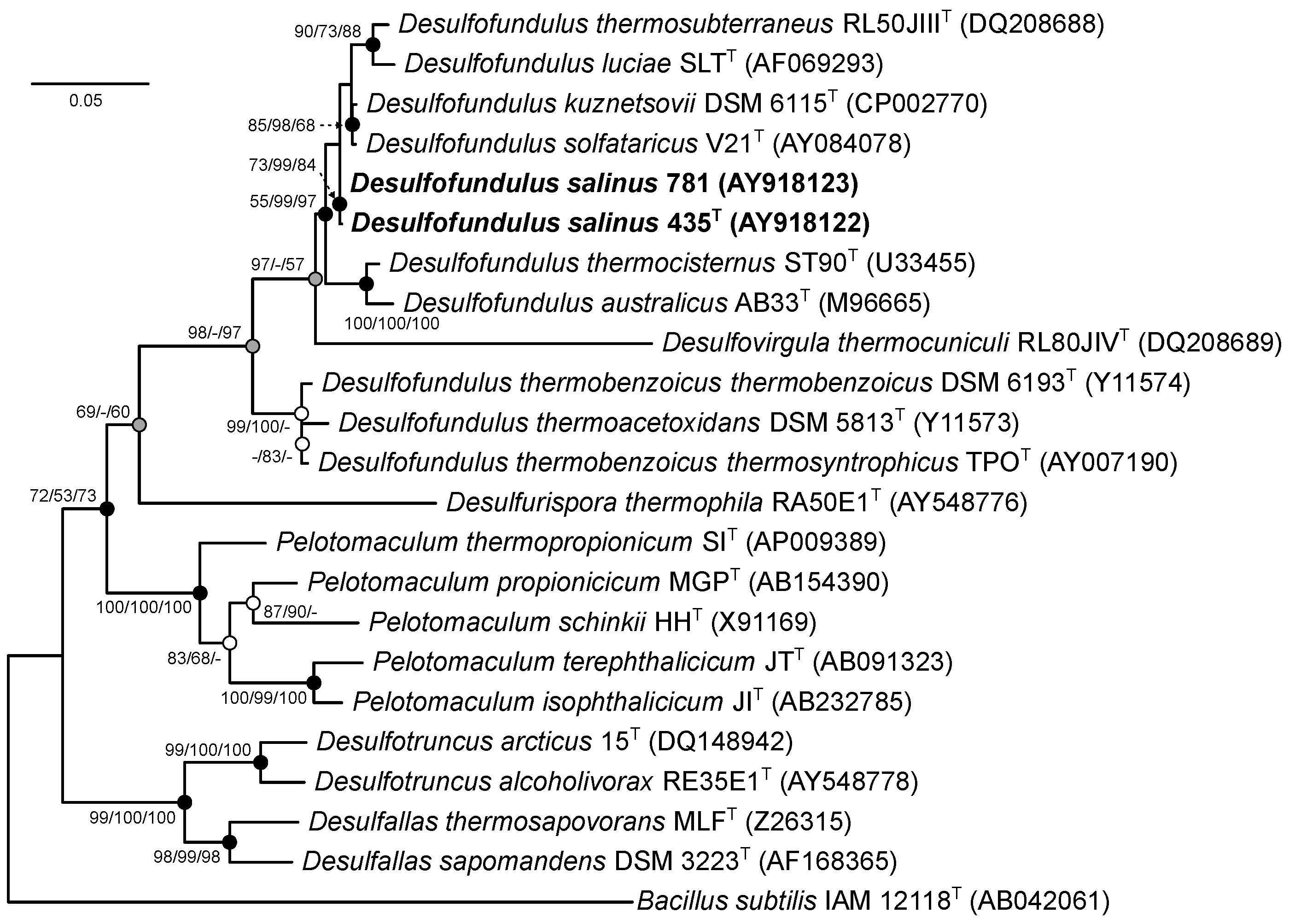
3.1. Phylogenetic Analysis of the 16S rRNA Gene
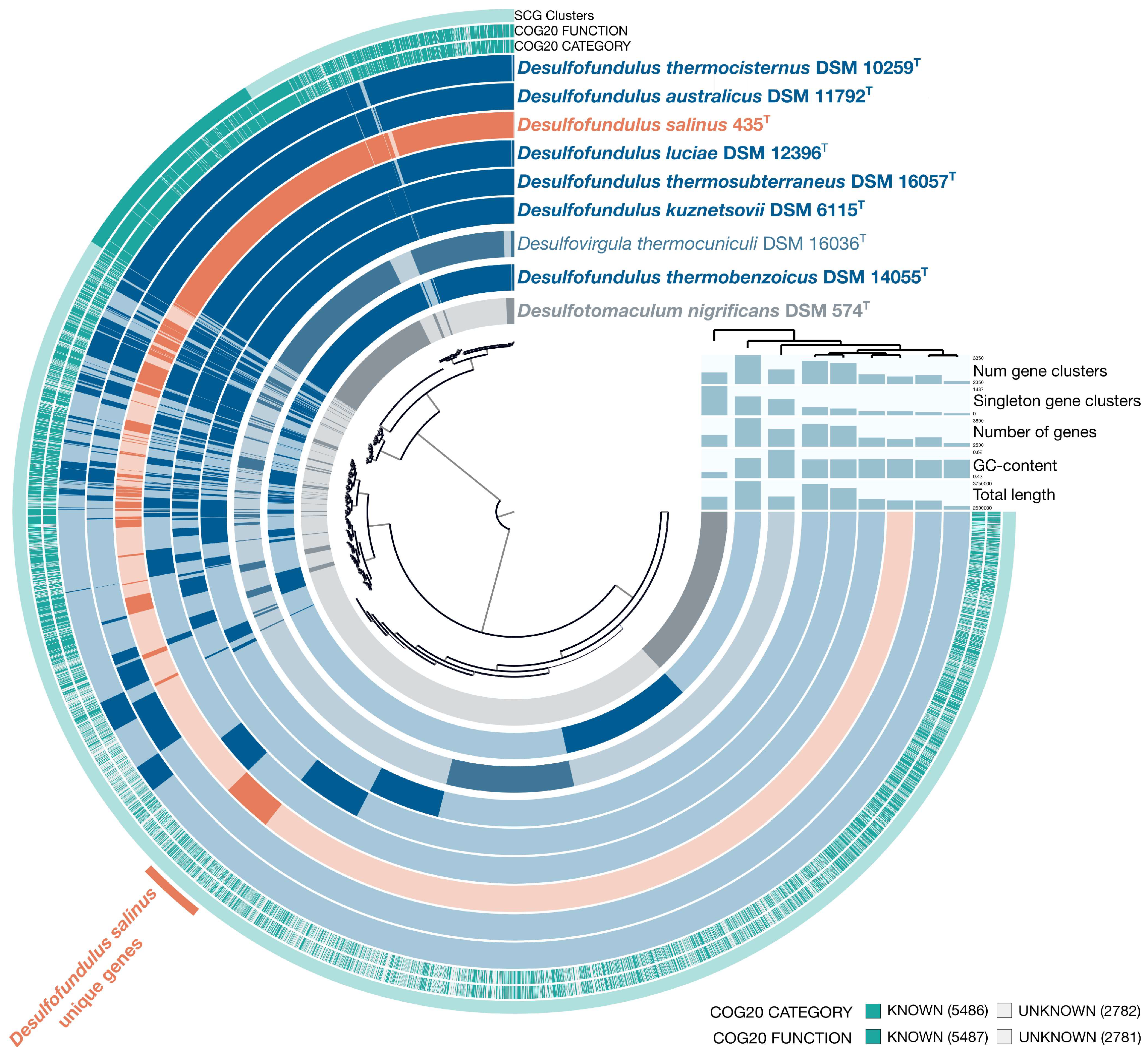
3.2. Genome Features and Phylogeny
3.3. Genome Insights
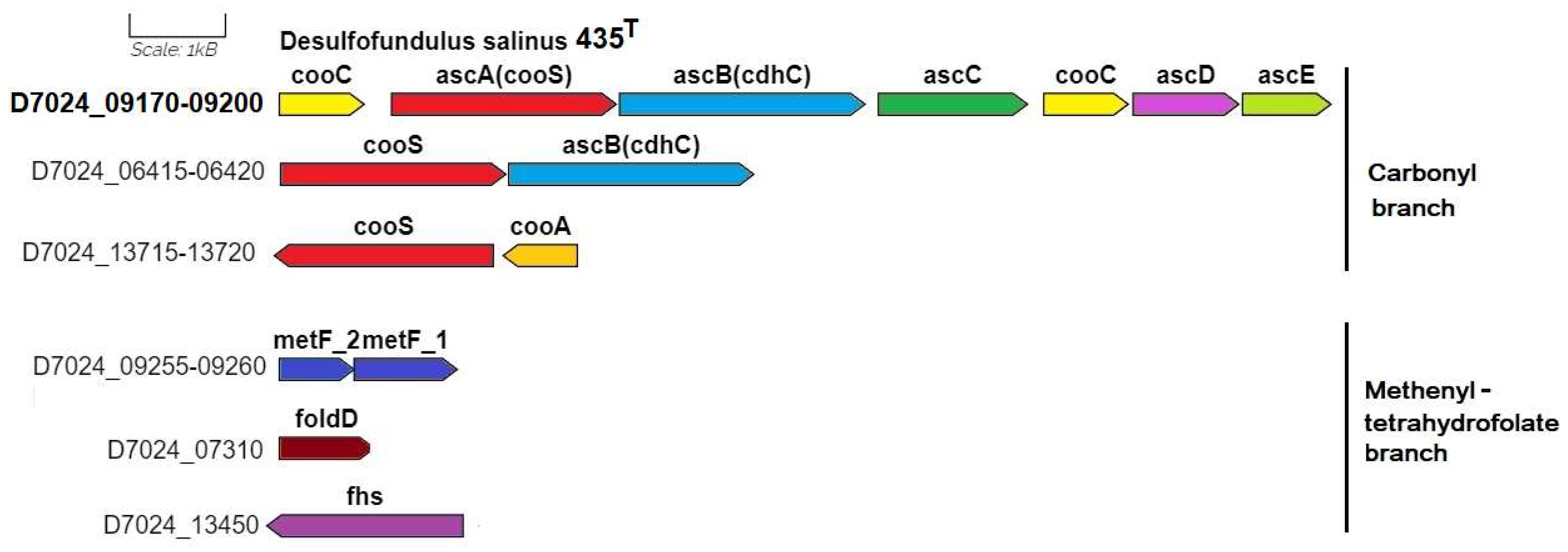
3.3.1. Autotrophic CO2 Fixation
3.3.2. Sulfur Metabolism
3.3.3. Carbohydrate Metabolism and Oxidation of Organic Compounds
3.3.4. Methanol Metabolism
3.3.5. Methane Metabolism
3.3.6. Nitrogen Metabolism
3.3.7. Hydrogenase Genes
3.3.8. Sporulation Genes
3.4. Phenotypic Characteristics of Strains 435T and 781
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell, L.L.; Postgate, J.R. Classification of the spore-forming sulfate-reducing bacteria. Bacteriol. Rev. 1965, 29, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Sproer, C.; Rainey, F.A.; Burghardt, J.; Pauker, O.; Hippe, H. Phylogenetic analysis of the genus Desulfotomaculum: Evidence for the misclassification of Desulfotomaculum guttoideum and description of Desulfotomaculum orientis as Desulfosporosinus orientis gen. nov., comb. nov. Int. J. Syst. Bacteriol. 1997, 47, 1134–1139. [Google Scholar] [CrossRef]
- Watanabe, M.; Kojima, H.; Fukui, M. Review of Desulfotomaculum species and proposal of the genera Desulfallas gen. nov., Desulfofundulus gen. nov., Desulfofarcimen gen. nov. and Desulfohalotomaculum gen. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Rozanova, E.P. Thermophilic sulfate-reducing bacteria from oil strata. Mikrobiologiya 1978, 47, 142–148. (In Russian) [Google Scholar]
- Nazina, T.N.; Pivovarova, T.A. Submicroscopic organization and sporulation in Desulfotomaculum nigrificans. Microbiology 1979, 48, 241–246. [Google Scholar]
- Nazina, T.N.; Rozanova, E.P.; Kalininskaia, T.A. Fixation of molecular nitrogen by sulfate-reducing bacteria from petroleum strata. Mikrobiologiia 1979, 48, 133–136. (In Russian) [Google Scholar] [PubMed]
- Belyakova, E.V.; Rozanova, E.P. Newly discovered properties of spore-forming sulfate-reducing bacteria, Desulfotomaculum strains 435 and 781. Microbiology 2004, 73, 284–286. [Google Scholar] [CrossRef]
- Nazina, T.N.; Rozanova, E.P.; Belyakova, E.V.; Lysenko, A.M.; Poltaraus, A.B.; Tourova, T.P.; Osipov, G.A.; Belyaev, S.S. Description of “Desulfotomaculum nigrificans subsp. salinus” as a new species, Desulfotomaculum salinum sp. nov. Microbiology 2005, 74, 567–574. [Google Scholar] [CrossRef]
- Grouzdev, D.S.; Bidzhieva, S.K.; Tourova, T.P.; Krutkina, M.S.; Poltaraus, A.B.; Nazina, T.N. Draft genome sequence of a sulfate-reducing bacterium, ‘Desulfofundulus salinum’ 435T, isolated from a high-temperature gas field in Russia. Microbiol. Resour. Announc. 2018, 7, e01408-18. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Notification list. Notification that new names and new combinations have appeared in volume 68, part 9 of the IJSEM. Int. J. Syst. Evol. Microbiol. 2018, 68, 3685–3687. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E. The emended family Peptococcaceae and description of the families Desulfitobacteriaceae, Desulfotomaculaceae, and Thermincolaceae. In The Prokaryotes; Rosenberg, E., DeLong, E., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 286–290. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Ivanova, A.E.; Kanchaveli, L.P.; Rozanova, E.P. A new sporeforming thermophilic methylotrophic sulfate-reducing bacterium, Desulfotomaculum kuznetsovii sp. nov. Microbiology 1988, 57, 659–663. [Google Scholar]
- Validation of the Publication of New Names and New Combinations Previously Effectively Published Outside the IJSB: List No. 35. Int. J. Syst. Bacteriol. 1990, 40, 470–471. [CrossRef]
- Postgate, J.R. Media for sulfur bacteria. Lab. Pract. 1966, 15, 1239–1244. [Google Scholar] [PubMed]
- Widdel, F. The genus Desulfotomaculum. In The Prokaryotes, 2nd ed.; Balows, A., Trüper, H.G., Dworkin, M., Harder, W., Schleifer, K.H., Eds.; Springer: New York, NY, USA; Berlin/Heidelberg, Germany, 1992; Volume 1, pp. 1792–1799. [Google Scholar]
- Pfennig, N.; Lippert, K.D. Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch. Für Mikrobiol. 1966, 55, 245–256. [Google Scholar] [CrossRef]
- Cypionka, H.; Pfennig, N. Growth yields of Desulfotomaculum orientis with hydrogen in chemostat culture. Arch. Microbiol. 1986, 143, 366–369. [Google Scholar] [CrossRef]
- Trüper, H.G.; Schlegel, H.G. Sulfur metabolism in Thiorhodaceae. I. Quantitative measurements on growing cells of Chromatium okenii. Antonie Van Leeuwenhoek 1964, 30, 321–323. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Ciufo, S.; Li, W. Prokaryotic genome annotation pipeline. In The NCBI Handbook, 2nd ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2013. [Google Scholar]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Vinh, L.S.; Flouri, T.; Stamatakis, A.; von Haeseler, A.; Minh, B.Q. MPBoot: Fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol. Biol. 2018, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Delmont, T.O.; Eren, E.M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.J.; de Crécy-Lagard, V.; Zallot, R. Gene Graphics: A genomic neighborhood data visualization web application. Bioinformatics 2018, 34, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Pierce, E.; Xie, G.; Barabote, R.D.; Saunders, E.; Han, C.S.; Detter, J.C.; Richardson, P.; Brettin, T.S.; Das, A.; Ljungdahl, L.G.; et al. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ. Microbiol. 2008, 10, 2550–2573. [Google Scholar] [CrossRef] [PubMed]
- Parshina, S.N.; Sipma, J.; Nakashimada, Y.; Henstra, A.M.; Smidt, H.; Lysenko, A.M.; Lens, P.N.L.; Lettinga, G.; Stams, A.J.M. Desulfotomaculum carboxydivorans sp. nov.; a novel sulfate-reducing bacterium capable of growth at 100% CO. Int. J. Syst. Evol. Microbiol. 2005, 55, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Parshina, S.N.; Alves, J.I.; Sousa, D.Z.; Pereira, I.A.; Muyzer, G.; Kuever, J.; Lebedinsky, A.V.; Koehorst, J.J.; Worm, P.; et al. Genome analyses of the carboxydotrophic sulfate-reducers Desulfotomaculum nigrificans and Desulfotomaculum carboxydivorans and reclassification of Desulfotomaculum caboxydivorans as a later synonym of Desulfotomaculum nigrificans. Stand. Genom. Sci. 2014, 9, 655–675. [Google Scholar] [CrossRef]
- Spormann, A.M.; Thauer, R.K. Anaerobic acetate oxidation to CO2 in Desulfotomaculum acetoxidans. Demonstration of enzymes required for the operation of an oxidative acetyl-CoA/carbon monoxide dehydrogenase pathway. Arch. Microbiol. 1988, 150, 374–380. [Google Scholar] [CrossRef]
- Spormann, A.M.; Thauer, R.K. Anaerobic acetate oxidation to CO2 by Desulfotomaculum acetoxidans. Arch. Microbiol. 1989, 152, 189–195. [Google Scholar] [CrossRef]
- Spring, S.; Lapidus, A.; Schröder, M.; Gleim, D.; Sims, D.; Meincke, L.; Glavina Del Rio, T.; Tice, H.; Copeland, A.; Cheng, J.F.; et al. Complete genome sequence of Desulfotomaculum acetoxidans type strain (5575). Stand. Genom. Sci. 2009, 1, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Kuever, J.; Visser, M.; Loeffler, C.; Boll, M.; Worm, P.; Sousa, D.Z.; Plugge, C.M.; Schaap, P.J.; Muyzer, G.; Pereira, I.A.C.; et al. Genome analysis of Desulfotomaculum gibsoniae strain GrollT a highly versatile Gram-positive sulfate-reducing bacterium. Stand. Genom. Sci. 2014, 9, 821–839. [Google Scholar] [CrossRef] [PubMed]
- Krämer, M.; Cypionka, H. Sulfate formation via ATP sulfurylase in thiosulfate- and sulfite-disproportionating bacteria. Arch. Microbiol. 1989, 151, 232–237. [Google Scholar] [CrossRef]
- Jackson, B.E.; McInerney, M.J. Thiosulfate disproportionation by Desulfotomaculum thermobenzoicum. Appl. Environ. Microbiol. 2000, 66, 3650–3653. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Worm, P.; Muyzer, G.; Pereira, I.A.C.; Schaap, P.; Plugge, C.M.; Kuever, J.; Parshina, S.N.; Nazina, T.N.; Ivanova, A.E.; et al. Genome analysis of Desulfotomaculum kuznetsovii strain 17T reveals a physiological similarity with Pelotomaculum thermopropionicum strain SIT. Stand. Genom. Sci. 2013, 8, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.Z.; Visser, M.; van Gelder, A.H.; Boeren, S.; Pieterse, M.M.; Pinkse, M.W.H.; Verhaert, P.D.E.M.; Vogt, C.; Franke, S.; Kümmel, S.; et al. The deep-subsurface sulfate reducer Desulfotomaculum kuznetsovii employs two methanol-degrading pathways. Nat. Commun. 2018, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Friedeheim, L.; Boeren, S.; Sánchez-Andrea, I.; Stams, A.J.M.; Sousa, D.Z. Alcohol dehydrogenase system acts as the sole pathway for methanol oxidation in Desulfofundulus kuznetsovii strain TPOSR. Antonie Van Leeuwenhoek 2024, 117, 47. [Google Scholar] [CrossRef]
- Goorissen, H.P.; Stams, A.J.M.; Hansen, T.A. Methanol dissimilation in Desulfotomaculum kuznetsovii. In Thermophilic Methanol Utilization by Sulfate Reducing Bacteria; Ph.D. Dissertation; University of Groningen: Groningen, The Netherlands, 2002; Chapter 3; pp. 55–61. [Google Scholar]
- Copeland, A.; Spring, S.; Goker, M.; Schneider, S.; Lapidus, A.; Del Rio, T.G.; Tice, H.; Cheng, J.F.; Chen, F.; Nolan, M.; et al. Complete genome sequence of Desulfomicrobium baculatum type strain (X). Stand. Genom. Sci. 2009, 1, 29–37. [Google Scholar] [CrossRef]
- Postgate, J.R. Nitrogen fixation by sporulating sulphate reducing bacteria including rumen strains. J. Gen. Microbiol. 1970, 63, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.C.; Fang, Z.; Mason, S.W.; Setubal, J.C.; Dixon, R. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genom. 2012, 13, 162. [Google Scholar] [CrossRef]
- Taylor, B.L.; Zhulin, I.B. PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef] [PubMed]
- Schut, G.J.; Adams, M.W. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: A new perspective on anaerobic hydrogen production. J. Bacteriol. 2009, 191, 4451–4457. [Google Scholar] [CrossRef] [PubMed]
- Malki, S.; Saimmaime, I.; De Luca, G.; Rousset, M.; Dermoun, Z.; Belaich, J.P. Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 1995, 177, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Böck, A.; King, P.W.; Blokesch, M.; Posewitz, M.C. Maturation of Hydrogenases. Adv. Microb. Physiol. 2006, 51, 1–225. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vecchia, E.; Visser, M.; Stams, A.J.M.; Bernier-Latmani, R. Sporulation in the Desulfotomaculum genus. Environ. Microbiol. Rep. 2014, 6, 756–766. [Google Scholar] [CrossRef]
- Khanna, K.; Lopez-Garrido, J.; Pogliano, K. Shaping an endospore: Architectural transformations during Bacillus subtilis sporulation. Annu. Rev. Microbiol. 2020, 74, 361–386. [Google Scholar] [CrossRef]
- Khanna, K.; Lopez-Garrido, J.; Sugie, J.; Pogliano, K.; Villa, E. Asymmetric localization of the cell division machinery during Bacillus subtilis sporulation. Elife 2021, 10, e62204. [Google Scholar] [CrossRef]
- Love, A.C.; Patel, B.K.; Nichols, P.D.; Stackebrandt, E. Desulfotomaculum australicum, sp. nov., a thermophilic sulfate-reducing bacterium isolated from the Great Artesian Basin in Australia. Syst. Appl. Microbiol. 1993, 16, 244–251. [Google Scholar] [CrossRef]
- Nilsen, R.K.; Torsvik, T.; Lien, T. Desulfotomaculum thermocisternum sp. nov., a sulfate reducer isolated from a hot North Sea oil reservoir. Int. J. Syst. Bacteriol. 1996, 46, 397–402. [Google Scholar] [CrossRef]
- Liu, V.; Karnauchow, T.M.; Jarrell, K.F.; Balkwill, D.L.; Drake, G.R.; Ringelberg, D.; Clarno, R.; Boone, D.R. Description of two new thermophilic Desulfotomaculum spp., Desulfotomaculum putei sp. nov., from a deep terrestrial subsurface, and Desulfotomaculum luciae sp. nov., from a hot spring. Int. J. Syst. Bacteriol. 1997, 47, 615–621. [Google Scholar] [CrossRef]
- Goorissen, H.P.; Boschker, T.S.; Stams, A.J.M.; Hansen, T.A. Isolation of thermophilic Desulfotomaculum strains with methanol and sulfite from solfataric mud pools, and characterization of Desulfotomaculum solfataricum sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, A.H.; Spring, S.; Schumann, P.; Kroppenstedt, R.M.; Puhakka, J.A. Desulfotomaculum thermosubterraneum sp. nov., a thermophilic sulfate-reducer isolated from an underground mine located in a geothermally active area. Int. J. Syst. Evol. Microbiol. 2006, 56, 2603–2608. [Google Scholar] [CrossRef]
- Min, H.; Zinder, S.H. Isolation and characterization of a thermophilic sulfate-reducing bacterium Desulfotomaculum thermoacetoxidans sp. nov. Arch. Microbiol. 1990, 153, 399–404. [Google Scholar] [CrossRef]
- Tasaki, M.; Kamagata, Y.; Nakamura, K.; Mikami, E. Isolation and characterization of a thermophilic benzoate-degrading, sulfate-reducing bacterium, Desulfotomaculum thermobenzoicum sp. nov. Arch. Microbiol. 1991, 155, 348–352. [Google Scholar] [CrossRef]
- Plugge, C.M.; Balk, M.; Stams, A.J.M. Desulfotomaculum thermobenzoicum subsp. thermosyntrophicum subsp. nov., a thermophilic, syntrophic, propionate-oxidizing, spore-forming bacterium. Int. J. Syst. Evol. Microbiol. 2002, 52, 391–399. [Google Scholar] [CrossRef]
- Nakagawa, S.; Inagaki, F.; Suzuki, Y.; Steinsbu, B.O.; Lever, M.A.; Takai, K.; Engelen, B.; Sako, Y.; Wheat, C.G.; Horikoshi, K. Microbial community in black rust exposed to hot ridge flank crustal fluids. Appl. Environ. Microbiol. 2006, 72, 6789–6799. [Google Scholar] [CrossRef]
- Frank, Y.A.; Kadnikov, V.V.; Gavrilov, S.N.; Banks, D.; Gerasimchuk, A.L.; Podosokorskaya, O.A.; Merkel, A.Y.; Chernyh, N.A.; Mardanov, A.V.; Ravin, N.V.; et al. Stable and variable parts of microbial community in Siberian deep subsurface thermal aquifer system revealed in a long-term monitoring study. Front. Microbiol. 2016, 7, 2101. [Google Scholar] [CrossRef]
- Aüllo, T.; Ranchou-Peyruse, A.; Ollivier, B.; Magot, M. Desulfotomaculum spp. and related gram-positive sulfate-reducing bacteria in deep subsurface environments. Front. Microbiol. 2013, 4, 362. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type Strain | Genome | Genome Size, Mb | Genomic G + C Content, mol.% | Strain 435T | ||
|---|---|---|---|---|---|---|
| 16S rRNA | dDDH | ANI | ||||
| Strain 435T | GCF_003627965.1 | 2.88 | 55.1 | 100.0 | 100.0 | 100.0 |
| D. australicus AB33T | GCF_900129285 | 2.88 | 54.6 | 93.6 | 27.0 | 83.0 |
| D. kuznetsovii 17T | GCF_000214705 | 3.60 | 54.9 | 98.0 | 51.1 | 92.7 |
| D. luciae SLT T | GCF_030813795 | 2.95 | 55.2 | 94.7 | 52.2 | 93.3 |
| D. thermocisternus ST90T | GCF_000686645 | 2.65 | 54.7 | 97.5 | 27.8 | 83.4 |
| D. thermosubterraneus RL50JIIIT | GCF_900142025 | 3.41 | 54.9 | 95.6 | 47.5 | 92.1 |
| D. solfataricus V21T | ND * | ND | ND | 98.4 | ND | ND |
| D. thermoacetoxidans CAMZT | ND | ND | ND | 94.3 | ND | ND |
| D. thermobenzoicus subsp. thermobenzoicus TSBT | ND | ND | ND | 92.2 | ND | ND |
| D. thermobenzoicus subsp. thermosyntrophicus TPO | GCF_009428905 | 3.71 | 55.8 | 95.2 | 25.0 | 78.7 |
| Characteristic | Strain, Species | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Motility | + | + | + | + | + | + | – | + | + | + | + |
| Temperature range, °C | 45–70 | 45–70 | 50–85 | 40–74 | 41–75 | 50–70 | 48–65 | 50–72 | 45–65 | 40–70 | 45–62 |
| Temperature optimum, °C | 55–60 | 55–60 | 60–65 | 68 | 62 | 60–65 | 60 | 61–66 | 55–60 | 62 | 55 |
| NaCl range/optimum, % (w/v) | 0–3/ 1 | 0–4.5/ 0.5 | 0–3/ 0 | >0/ ND | 0–4.7/ 0.3–1.2 | 0–3/ <1 | 6.4–7.9/7.3 | 0–1.5/ 0–1 | 0–1.5/ 0 | 0/ 0 | ND |
| pH range | 6.0–8.5 | 6.0–8.5 | ND | 5.5–8.5 | 6.2–8.9 | 6.3–8.3 | 0–1.5 | 6.4–7.8 | 6.0–7.5 | 6–8 | 6–8 |
| pH optimum | 7.0–8.0 | 7.0–8.0 | 7.0–7.2 | 7–7.4 | 6.7 | 7.0–7.9 | 0 | 7.2–7.4 | 6.5 | 7.2 | 7.0 |
| Electron donors with sulfate: | |||||||||||
| Formate | + | + | + | – | – | + | + | + | + | + | ND |
| Acetate | – | – | + | + | – | – | + | – | + | + | – |
| Propionate | + | + | + | – | + | – | + | + | + | + | + |
| Butyrate | + | + | + | – | + | – | + | + | + | + | – |
| Valerate | + | + | + | + | ND | + | + | ND | |||
| Hexadecanoate | + | + | + | + | + | ND | + | + | – | ND | ND |
| Fumarate | + | + | + | – | ND | + | + | ND | + | ND | |
| Malate | + | + | + | – | – | + | + | + | + | – | |
| Succinate | + | + | + | – | – | + | + | + | – | – | |
| Benzoate | – | – | – | + | – | ND | – | – | – | + | + |
| Methanol | + | + | + | – | – | – | + | – | ND | – | – |
| Ethanol | + | + | + | + | + | + | + | + | – | + | – |
| Propanol | + | + | + | ND | + | ND | + | + | + | + | – |
| Butanol | + | + | + | ND | + | – | + | + | + | + | – |
| Glucose | – | – | – | – | – | +W | – | – | – | – | |
| Fructose | – | – | – | ND | – | – | + | – | ND | – | – |
| L-Alanine | – | – | – | ND | ND | – | + | + | ND | ND | |
| Electron acceptors: | |||||||||||
| Thiosulfate | + | + | + | ND | + | + | + | + | + | + | – |
| Sulfite | + | + | + | ND | + | +– | + | + | – | + | – |
| Sulfur | – | – | – | ND | – | + | + | + | + | – | ND |
| Nitrate | – | – | – | ND | – | – | – | – | – | + | – |
| Lactate fermentation | – | – | + | + | ND | + | + | + | – | + | + |
| Parameter | Desulfofundulus salinus sp. nov. |
|---|---|
| Genus name | Desulfofundulus |
| Species name | Desulfofundulus salinus |
| Species status | sp. nov. |
| Species etymology | sa.li’nus. N.L. masc. adj. salinus, salty |
| Designation of the Type Strain | 435T |
| Strain Collection Numbers | VKM B-1492T = DSM 23196T |
| Genome accession number | GCF_003627965.1 |
| Genome status | Incomplete |
| Genome size | 2886 kbp |
| GC mol% | 55.1 |
| 16S rRNA gene accession nr. | AY918122 |
| Description of the new taxon and diagnostic traits | The cells are rod-shaped or spindle-shaped (lemon-shaped), motile due to peritrichous flagella, stained Gram-negative, but have cell wall structure typical of Gram-positive bacteria. Endospores are spherical, located centrally or subterminally and slightly distend the cells. Growth is observed in the presence of 0–4.5% (w/v) NaCl (optimum, 0.5–1% NaCl), at a pH of 6.0–8.5 (optimum, pH 7.0), and at 45–70 °C (optimum, 55–60 °C) in a sulfate-reducing condition. Strictly anaerobic. Reduces sulfate to sulfide in media with H2/CO2, formate, lactate, pyruvate, malate, fumarate, succinate, methanol, ethanol, propanol, butanol, propionate, butyrate, valerate, and palmitate as carbon and energy sources, but does not use L-alanine, L-serine, L-arginine, L-cysteine, glucose, fructose, lactose, benzoate, citrate, tartrate, glycerol, glutamate, threonine, tryptophan, asparagine, glutamate, and phenylalanine. Lactate is oxidized with the production of acetate. No vitamins or other growth factors are required, although the addition of yeast extract stimulates growth. Pyruvate and (weakly) fumarate are used for fermentative growth, but lactate is not fermented. Utilizes sulfate, thiosulfate, and sulfite as electron acceptors in the presence of lactate, but does not use nitrate. The predominant cellular fatty acids are iso-C15:0, iso-C17:0, C16:0, and C18:0. The genome size of the type strain is 2.886 Mb with a genomic G + C content of 55.1 mol%. The type strain, 435T (=VKM B-1492T =DSM 23196T), was isolated from the Igrim gas field, in Khanty-Mansiysk Autonomous Okrug, Yugra, Russian Federation. The GenBank/EMBL/DDBJ accession number for the 16S rRNA gene sequence is AY918122 and the genomic assembly accession number is GCF_003627965.1. The reference strain is 781 (VKM B-1379). |
| Country and region of origin | Russian Federation, Khanty-Mansiysk Autonomous Okrug, Yugra |
| Date of isolation | 1973 |
| Source of isolation | A mixture of condensation and reservoir water from the Igrim gas field |
| Sampling date | 1973 |
| Latitude, Longitude | 63°06′2.12″ N, 64°19′54.15″ E |
| Depth (meters below sea level) | 1600–1620 |
| Number of strains in study | 2 |
| Source of non-type strain | Strain 781 was isolated from the Ust-Balyk oil and gas field, Russia |
| Information related to the Nagoya Protocol | Not applicable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazina, T.N.; Tourova, T.P.; Grouzdev, D.S.; Bidzhieva, S.K.; Poltaraus, A.B. A Novel View on the Taxonomy of Sulfate-Reducing Bacterium ‘Desulfotomaculum salinum’ and a Description of a New Species Desulfofundulus salinus sp. nov. Microorganisms 2024, 12, 1115. https://doi.org/10.3390/microorganisms12061115
Nazina TN, Tourova TP, Grouzdev DS, Bidzhieva SK, Poltaraus AB. A Novel View on the Taxonomy of Sulfate-Reducing Bacterium ‘Desulfotomaculum salinum’ and a Description of a New Species Desulfofundulus salinus sp. nov. Microorganisms. 2024; 12(6):1115. https://doi.org/10.3390/microorganisms12061115
Chicago/Turabian StyleNazina, Tamara N., Tatyana P. Tourova, Denis S. Grouzdev, Salimat K. Bidzhieva, and Andrey B. Poltaraus. 2024. "A Novel View on the Taxonomy of Sulfate-Reducing Bacterium ‘Desulfotomaculum salinum’ and a Description of a New Species Desulfofundulus salinus sp. nov." Microorganisms 12, no. 6: 1115. https://doi.org/10.3390/microorganisms12061115