
Contribution of Toxin–Antitoxin Systems to Adherent-Invasive E. coli Pathogenesis


Abstract
1. Introduction
2. Crohn’s Disease and Adherent-Invasive Escherichia coli
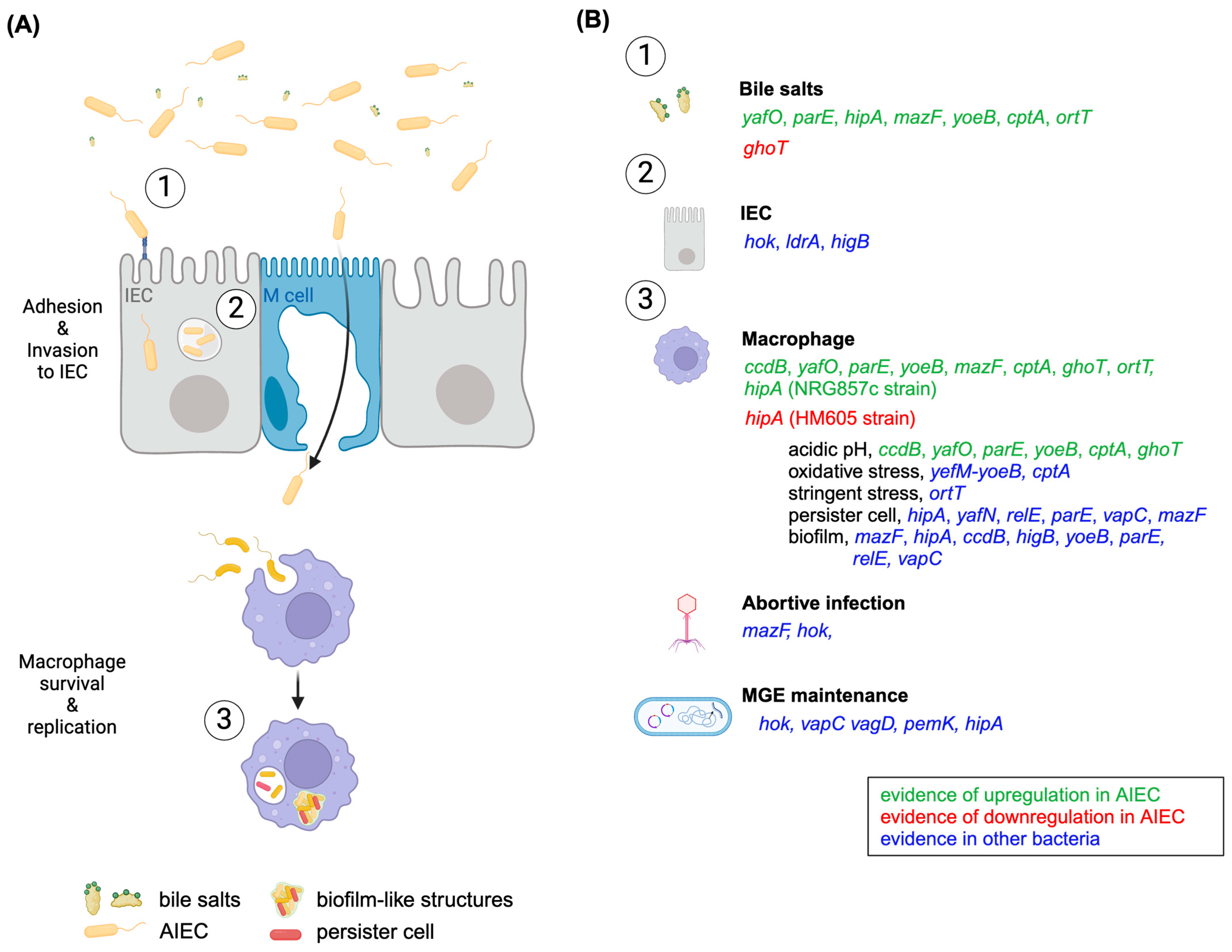
3. Putative TA Roles in AIEC’s Pathogenesis
3.1. Roles in Stress Response
3.2. Roles in Biofilm Formation
3.3. Role as Phage Inhibition Systems
3.4. Roles in MGEs Maintenance
3.5. Role in the Host Lifestyle
3.5.1. Intra-IECs Lifestyle
3.5.2. Intra-Macrophage Lifestyle
3.6. Roles in Persister Cell Formation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic Toxin–Antitoxin Stress Response Loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef]
- Jurėnas, D.; Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Biology and Evolution of Bacterial Toxin–Antitoxin Systems. Nat. Rev. Microbiol. 2022, 20, 335–350. [Google Scholar] [CrossRef]
- Równicki, M.; Lasek, R.; Trylska, J.; Bartosik, D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins 2020, 12, 568. [Google Scholar] [CrossRef]
- Gerdes, K.; Rasmussen, P.B.; Molin, S. Unique Type of Plasmid Maintenance Function: Postsegregational Killing of Plasmid-Free Cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3116–3120. [Google Scholar] [CrossRef]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II Toxin-Antitoxin Systems: Evolution and Revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef]
- Wang, X.; Yao, J.; Sun, Y.-C.; Wood, T.K. Type VII Toxin/Antitoxin Classification System for Antitoxins That Enzymatically Neutralize Toxins. Trends Microbiol. 2021, 29, 388–393. [Google Scholar] [CrossRef]
- Choi, J.S.; Kim, W.; Suk, S.; Park, H.; Bak, G.; Yoon, J.; Lee, Y. The Small RNA, SdsR, Acts as a Novel Type of Toxin in Escherichia coli. RNA Biol. 2018, 15, 1319–1335. [Google Scholar] [CrossRef]
- Norton, J.P.; Mulvey, M.A. Toxin-Antitoxin Systems Are Important for Niche-Specific Colonization and Stress Resistance of Uropathogenic Escherichia coli. PLoS Pathog. 2012, 8, e1002954. [Google Scholar] [CrossRef]
- Bustamante, P.; Vidal, R. Repertoire and Diversity of Toxin—Antitoxin Systems of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. New Insight of T His Emergent E. coli Pathotype. Front. Microbiol. 2020, 11, 807. [Google Scholar] [CrossRef]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-Antitoxins and Bacterial Virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory Bowel Disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of Disease: Pathogenesis of Crohn’s Disease and Ulcerative Colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 73992. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.-L.; Barnich, N.; Bringer, M.-A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.-F. High Prevalence of Adherent-Invasive Escherichia coli Associated with Ileal Mucosa in Crohn’s Disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- López-Siles, M.; Camprubí-Font, C.; Gómez del Pulgar, E.M.; Sabat Mir, M.; Busquets, D.; Sanz, Y.; Martinez-Medina, M. Prevalence, Abundance, and Virulence of Adherent-Invasive Escherichia coli in Ulcerative Colitis, Colorectal Cancer, and Coeliac Disease. Front. Immunol. 2022, 13, 748839. [Google Scholar] [CrossRef] [PubMed]
- Kittana, H.; Gomes-Neto, J.C.; Heck, K.; Juritsch, A.F.; Sughroue, J.; Xian, Y.; Mantz, S.; Segura Muñoz, R.R.; Cody, L.A.; Schmaltz, R.J.; et al. Evidence for a Causal Role for Escherichia coli Strains Identified as Adherent-Invasive (AIEC) in Intestinal Inflammation. mSphere 2023, 8, e0047822. [Google Scholar] [CrossRef] [PubMed]
- Nash, J.H.; Villegas, A.; Kropinski, A.M.; Aguilar-Valenzuela, R.; Konczy, P.; Mascarenhas, M.; Ziebell, K.; Torres, A.G.; Karmali, M.A.; Coombes, B.K. Genome Sequence of Adherent-Invasive Escherichia coli and Comparative Genomic Analysis with Other E. coli Pathotypes. BMC Genom. 2010, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Boudeau, J.; Glasser, A.-L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive Ability of an Escherichia coli Strain Isolated from the Ileal Mucosa of a Patient with Crohn’s Disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [CrossRef] [PubMed]
- Glasser, A.-L.; Boudeau, J.; Barnich, N.; Perruchot, M.-H.; Colombel, J.-F.; Darfeuille-Michaud, A. Adherent Invasive Escherichia coli Strains from Patients with Crohn’s Disease Survive and Replicate within Macrophages without Inducing Host Cell Death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef]
- Gibold, L.; Garenaux, E.; Dalmasso, G.; Gallucci, C.; Cia, D.; Mottet-Auselo, B.; Faïs, T.; Darfeuille-Michaud, A.; Nguyen, H.T.T.; Barnich, N.; et al. The Vat-AIEC Protease Promotes Crossing of the Intestinal Mucus Layer by Crohn’s Disease-Associated Escherichia coli. Cell Microbiol. 2016, 18, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, E.; Chassaing, B.; Buisson, A.; Dubois, A.; de Vallée, A.; Billard, E.; Neut, C.; Bommelaer, G.; Colombel, J.-F.; Barnich, N.; et al. GipA Factor Supports Colonization of Peyer’s Patches by Crohn’s Disease-Associated Escherichia coli. Inflamm. Bowel. Dis. 2016, 22, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Delmas, J.; Gibold, L.; Faïs, T.; Batista, S.; Leremboure, M.; Sinel, C.; Vazeille, E.; Cattoir, V.; Buisson, A.; Barnich, N.; et al. Metabolic Adaptation of Adherent-Invasive Escherichia coli to Exposure to Bile Salts. Sci. Rep. 2019, 9, 2175. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Suzuki, H.; Herlekar, D.; Sartor, B.R.B.; Campbell, B.J.; Roberts, C.L.; Stewart, K.; Scherl, E.J.; Araz, Y.; Bitar, P.P.; et al. Inflammation-Associated Adherent-Invasive Escherichia coli Are Enriched in Pathways for Use of Propanediol and Iron and M-Cell Translocation. Inflamm. Bowel. Dis. 2014, 20, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Peyretaillade, E.; Claret, L.; de Vallée, A.; Dossat, C.; Vacherie, B.; Zineb, E.H.; Segurens, B.; Barbe, V.; Sauvanet, P.; et al. Complete Genome Sequence of Crohn’s Disease-Associated Adherent-Invasive E. coli Strain LF82. PLoS ONE 2010, 5, e12714. [Google Scholar] [CrossRef] [PubMed]
- Viladomiu, M.; Metz, M.L.; Lima, S.F.; Jin, W.B.; Chou, L.; Guo, C.J.; Diehl, G.E.; Simpson, K.W.; Scherl, E.J.; Longman, R.S. Adherent-Invasive E. coli Metabolism of Propanediol in Crohn’s Disease Regulates Phagocytes to Drive Intestinal Inflammation. Cell Host Microbe 2021, 29, 607–619.e8. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Alteri, C.J.; Rodrigues, M.; Nagao-Kitamoto, H.; Sugihara, K.; Himpsl, S.D.; Bazzi, M.; Miyoshi, M.; Nishioka, T.; Hayashi, A.; et al. Dietary L-Serine Confers a Competitive Fitness Advantage to Enterobacteriaceae in the Inflamed Gut. Nat. Microbiol. 2019, 5, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Fornelos, N.; Franzosa, E.A.; Bishai, J.; Annand, J.W.; Oka, A.; Lloyd-Price, J.; Arthur, T.D.; Garner, A.; Avila-Pacheco, J.; Haiser, H.J.; et al. Growth Effects of N-Acylethanolamines on Gut Bacteria Reflect Altered Bacterial Abundances in Inflammatory Bowel Disease. Nat. Microbiol. 2020, 5, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Barnich, N.; Carvalho, F.A.; Glasser, A.L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.F.; et al. CEACAM6 Acts as a Receptor for Adherent-Invasive E. coli, Supporting Ileal Mucosa Colonization in Crohn Disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef]
- Dreux, N.; Denizot, J.; Martinez-Medina, M.; Mellmann, A.; Billig, M.; Kisiela, D.; Chattopadhyay, S.; Sokurenko, E.; Neut, C.; Gower-Rousseau, C.; et al. Point Mutations in FimH Adhesin of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Enhance Intestinal Inflammatory Response. PLoS Pathog. 2013, 9, e1003141. [Google Scholar] [CrossRef]
- Rolhion, N.; Carvalho, F.A.; Darfeuille-Michaud, A. OmpC and the ΣE Regulatory Pathway Are Involved in Adhesion and Invasion of the Crohn’s Disease-Associated Escherichia coli Strain LF82. Mol. Microbiol. 2007, 63, 1684–1700. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Tran, H.T.; Lee, I.A.; Dreux, N.; Kamba, A.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-Binding Domains of Escherichia coli ChiA Mediate Interactions with Intestinal Epithelial Cells in Mice with Colitis. Gastroenterology 2013, 145, 602–612.e9. [Google Scholar] [CrossRef]
- Sevrin, G.; Massier, S.; Chassaing, B.; Agus, A.; Delmas, J.; Denizot, J.; Billard, E.; Barnich, N. Adaptation of Adherent-Invasive E. coli to Gut Environment: Impact on Flagellum Expression and Bacterial Colonization Ability. Gut. Microbes 2020, 11, 364–380. [Google Scholar] [CrossRef] [PubMed]
- Bringer, M.-A.; Barnich, N.; Glasser, A.-L.; Bardot, O.; Darfeuille-Michaud, A. HtrA Stress Protein Is Involved in Intramacrophagic Replication of Adherent and Invasive Escherichia coli Strain LF82 Isolated from a Patient with Crohn’s Disease. Infect. Immun. 2005, 73, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Bringer, M.A.; Rolhion, N.; Glasser, A.L.; Darfeuille-Michaud, A. The Oxidoreductase DsbA Plays a Key Role in the Ability of the Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Strain LF82 to Resist Macrophage Killing. J. Bacteriol. 2007, 189, 4860–4871. [Google Scholar] [CrossRef]
- Cieza, R.J.; Hu, J.; Ross, B.N.; Sbrana, E.; Torres, A.G. The IbeA Invasin of Adherent-Invasive Escherichia coli Mediates Interaction with Intestinal Epithelia and Macrophages. Infect. Immun. 2015, 83, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, K.T.; Nielsen, G.; Bjerrum, J.V.; Kruse, T.; Kallipolitis, B.H.; Møller-Jensen, J. A Role for the RNA Chaperone Hfq in Controlling Adherent-Invasive Escherichia coli Colonization and Virulence. PLoS ONE 2011, 6, e16387. [Google Scholar] [CrossRef] [PubMed]
- Demarre, G.; Prudent, V.; Schenk, H.; Rousseau, E.; Bringer, M.A.; Barnich, N.; Van Nhieu, G.T.; Rimsky, S.; De Monte, S.; Espéli, O. The Crohn’s Disease-Associated Escherichia coli Strain LF82 Relies on SOS and Stringent Responses to Survive, Multiply and Tolerate Antibiotics within Macrophages. PLoS Pathog 2019, 15, e1008123. [Google Scholar] [CrossRef] [PubMed]
- Prudent, V.; Demarre, G.; Vazeille, E.; Wery, M.; Quenech’Du, N.; Ravet, A.; Dauverd-Girault, J.; van Dijk, E.; Bringer, M.A.; Descrimes, M.; et al. The Crohn’s Disease-Related Bacterial Strain LF82 Assembles Biofilm-like Communities to Protect Itself from Phagolysosomal Attack. Commun. Biol. 2021, 4, 627. [Google Scholar] [CrossRef]
- Guan, J.; Chen, Y.; Goh, Y.-X.; Wang, M.; Tai, C.; Deng, Z.; Song, J.; Ou, H.-Y. TADB 3.0: An Updated Database of Bacterial Toxin-Antitoxin Loci and Associated Mobile Genetic Elements. Nucleic Acids Res. 2024, 52, D784–D790. [Google Scholar] [CrossRef]
- Lobato-Márquez, D.; Moreno-Córdoba, I.; Figueroa, V.; Díaz-Orejas, R.; García-del Portillo, F. Distinct Type I and Type II Toxin-Antitoxin Modules Control Salmonella Lifestyle inside Eukaryotic Cells. Sci. Rep. 2015, 5, 9374. [Google Scholar] [CrossRef]
- Sistrunk, J.R.; Nickerson, K.P.; Chanin, R.B.; Rasko, D.A.; Faherty, C.S. Survival of the Fittest: How Bacterial Pathogens Utilize Bile To Enhance Infection. Clin. Microbiol. Rev. 2016, 29, 819–836. [Google Scholar] [CrossRef]
- Chassaing, B.; Etienne-Mesmin, L.; Bonnet, R.; Darfeuille-Michaud, A. Bile Salts Induce Long Polar Fimbriae Expression Favouring Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Interaction with Peyer’s Patches. Environ. Microbiol. 2013, 15, 355–371. [Google Scholar] [CrossRef]
- Clarke, D.J.; Chaudhuri, R.R.; Martin, H.M.; Campbell, B.J.; Rhodes, J.M.; Constantinidou, C.; Pallen, M.J.; Loman, N.J.; Cunningham, A.F.; Browning, D.F.; et al. Complete Genome Sequence of the Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Strain HM605. J. Bacteriol. 2011, 193, 4540. [Google Scholar] [CrossRef]
- Bernard, P.; Kézdy, K.E.; Van Melderen, L.; Steyaert, J.; Wyns, L.; Pato, M.L.; Higgins, P.N.; Couturier, M. The F Plasmid CcdB Protein Induces Efficient ATP-Dependent DNA Cleavage by Gyrase. J. Mol. Biol. 1993, 234, 534–541. [Google Scholar] [CrossRef]
- Ogura, T.; Hiraga, S. Mini-F Plasmid Genes That Couple Host Cell Division to Plasmid Proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef]
- Gupta, K.; Tripathi, A.; Sahu, A.; Varadarajan, R. Contribution of the Chromosomal CcdAB Operon to Bacterial Drug Tolerance. J. Bacteriol. 2017, 199, e00397-17. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Salmon, M.A.; van Melderen, L.; Bernard, P.; Couturier, M. F Plasmid CcdB Killer Protein CcdB Gene Mutants Coding for Non-cytotoxic Proteins Which Retain Their Regulatory Functions. Mol. Microbiol. 1995, 15, 1031–1037. [Google Scholar] [CrossRef]
- Gerdes, K. Type II Toxin-Antitoxins Loci: The RelBE Family. In Prokaryotic Toxin-Antitoxins; Springer: Berlin/Heidelberg, Germany, 2013; pp. 69–92. [Google Scholar]
- Christensen-Dalsgaard, M.; Jørgensen, M.G.; Gerdes, K. Three New RelE-Homologous MRNA Interferases of Escherichia coli Differentially Induced by Environmental Stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef]
- Zhang, Y.; Yamaguchi, Y.; Inouye, M. Characterization of YafO, an Escherichia coli Toxin. J. Biol. Chem. 2009, 284, 25522–25531. [Google Scholar] [CrossRef]
- Singletary, L.A.; Gibson, J.L.; Tanner, E.J.; McKenzie, G.J.; Lee, P.L.; Gonzalez, C.; Rosenberg, S.M. An SOS-Regulated Type 2 Toxin-Antitoxin System. J. Bacteriol. 2009, 191, 7456–7465. [Google Scholar] [CrossRef]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE Toxin Encoded by the Broad-Host-Range Plasmid RK2 Is an Inhibitor of Escherichia coli Gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef]
- Kamruzzaman, M.; Iredell, J. A ParDE-Family Toxin Antitoxin System in Major Resistance Plasmids of Enterobacteriaceae Confers Antibiotic and Heat Tolerance. Sci. Rep. 2019, 9, 9872. [Google Scholar] [CrossRef]
- Cai, T.; Zhao, Q.; Xiang, W.; Zhu, L.; Rao, Y.; Tang, J. HigBA Toxin–Antitoxin System of Weissella Cibaria Is Involved in Response to the Bile Salt Stress. J. Sci. Food Agric. 2022, 102, 6749–6756. [Google Scholar] [CrossRef]
- Kirkpatrick, C.L.; Martins, D.; Redder, P.; Frandi, A.; Mignolet, J.; Chapalay, J.B.; Chambon, M.; Turcatti, G.; Viollier, P.H. Growth Control Switch by a DNA-Damage-Inducible Toxin–Antitoxin System in Caulobacter Crescentus. Nat. Microbiol. 2016, 1, 16008. [Google Scholar] [CrossRef]
- Janssen, B.D.; Garza-Sánchez, F.; Hayes, C.S. YoeB Toxin Is Activated during Thermal Stress. Microbiologyopen 2015, 4, 682–697. [Google Scholar] [CrossRef]
- Chan, W.; Domenech, M.; Moreno-Córdoba, I.; Navarro-Martínez, V.; Nieto, C.; Moscoso, M.; García, E.; Espinosa, M. The Streptococcus Pneumoniae YefM-YoeB and RelBE Toxin-Antitoxin Operons Participate in Oxidative Stress and Biofilm Formation. Toxins 2018, 10, 378. [Google Scholar] [CrossRef]
- Ma, D.; Gu, H.; Shi, Y.; Huang, H.; Sun, D.; Hu, Y. Edwardsiella Piscicida YefM-YoeB: A Type II Toxin-Antitoxin System That Is Related to Antibiotic Resistance, Biofilm Formation, Serum Survival, and Host Infection. Front. Microbiol. 2021, 12, 646299. [Google Scholar] [CrossRef]
- Kwan, B.W.; Lord, D.M.; Peti, W.; Page, R.; Benedik, M.J.; Wood, T.K. The MqsR/MqsA Toxin/Antitoxin System Protects E Scherichia Coli during Bile Acid Stress. Environ. Microbiol. 2015, 17, 3168–3181. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin MqsA Helps Mediate the Bacterial General Stress Response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef]
- Fraikin, N.; Rousseau, C.J.; Goeders, N.; Van Melderen, L. Reassessing the Role of the Type II MqsRA Toxin-Antitoxin System in Stress Response and Biofilm Formation: MqsA Is Transcriptionally Uncoupled from MqsR. mBio 2019, 10, e02678-19. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Benedik, M.J.; Wood, T.K. Orphan Toxin OrtT (YdcX) of Escherichia coli Reduces Growth during the Stringent Response. Toxins 2015, 7, 299–321. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Tan, Q.; Awano, N.; Yamaguchi, Y.; Inouye, M. A Novel Membrane-Bound Toxin for Cell Division, CptA (YgfX), Inhibits Polymerization of Cytoskeleton Proteins, FtsZ and MreB, in Escherichia coli. FEMS Microbiol. Lett. 2012, 328, 174–181. [Google Scholar] [CrossRef]
- McNeil, M.B.; Iglesias-Cans, M.C.; Clulow, J.S.; Fineran, P.C. YgfX (CptA) Is a Multimeric Membrane Protein That Interacts with the Succinate Dehydrogenase Assembly Factor SdhE (YgfY). Microbiology 2013, 159, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-X.; Zheng, K.-L.; Tang, A.-G.; Hu, X.-X.; Guo, X.-X.; Wu, C.; Cheng, Y.-Y. YgfY Contributes to Stress Tolerance in Shewanella Oneidensis Neither as an Antitoxin Nor as a Flavinylation Factor of Succinate Dehydrogenase. Microorganisms 2021, 9, 2316. [Google Scholar] [CrossRef] [PubMed]
- ElBanna, S.A.; Moneib, N.A.; Aziz, R.K.; Samir, R. Genomics-Guided Identification of a Conserved CptBA-like Toxin-Antitoxin System in Acinetobacter Baumannii. J. Adv. Res. 2021, 30, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Vestby, L.K.; Grønseth, T.; Simm, R.; Nesse, L.L. Bacterial Biofilm and Its Role in the Pathogenesis of Disease. Antibiotics 2020, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Medina, M.; Naves, P.; Blanco, J.; Aldeguer, X.; Blanco, J.E.; Blanco, M.; Ponte, C.; Soriano, F.; Darfeuille-Michaud, A.; Garcia-Gil, L.J. Biofilm Formation as a Novel Phenotypic Feature of Adherent-Invasive Escherichia coli(AIEC). BMC Microbiol. 2009, 9, 202. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Hoeflich, K.P.; Ikura, M.; Qing, G.; Inouye, M. MazF Cleaves Cellular MRNAs Specifically at ACA to Block Protein Synthesis in Escherichia coli. Mol. Cell. 2003, 12, 913–923. [Google Scholar] [CrossRef]
- Kolodkin-Gal, I.; Verdiger, R.; Shlosberg-Fedida, A.; Engelberg-Kulka, H. A Differential Effect of E. Coli Toxin-Antitoxin Systems on Cell Death in Liquid Media and Biofilm Formation. PLoS ONE 2009, 4, e6785. [Google Scholar] [CrossRef]
- Ma, D.; Mandell, J.B.; Donegan, N.P.; Cheung, A.L.; Ma, W.; Rothenberger, S.; Shanks, R.M.Q.; Richardson, A.R.; Urish, K.L. The Toxin-Antitoxin MazEF Drives Staphylococcus Aureus Biofilm Formation, Antibiotic Tolerance, and Chronic Infection. mBio 2019, 10, e01658-19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, X.; Ma, Q.; Zhang, X.-S.; Wood, T.K. Toxin-Antitoxin Systems in Escherichia coli Influence Biofilm Formation through YjgK (TabA) and Fimbriae. J. Bacteriol. 2009, 191, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The Bacterial Toxin RelE Displays Codon-Specific Cleavage of MRNAs in the Ribosomal A Site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef]
- Kasari, V.; Kurg, K.; Margus, T.; Tenson, T.; Kaldalu, N. The Escherichia coli MqsR and YgiT Genes Encode a New Toxin-Antitoxin Pair. J. Bacteriol. 2010, 192, 2908–2919. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Hay, A.J.; Zhong, Z.; Zhu, J.; Kan, B. Functional RelBE-Family Toxin-Antitoxin Pairs Affect Biofilm Maturation and Intestine Colonization in Vibrio Cholerae. PLoS ONE 2015, 10, e0135696. [Google Scholar] [CrossRef] [PubMed]
- Lemos, J.A.C.; Brown, T.A.; Abranches, J.; Burne, R.A. Characteristics of Streptococcus Mutans Strains Lacking the MazEF and RelBE Toxinâ-Antitoxin Modules. FEMS Microbiol. Lett. 2005, 253, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.; Sass, A.; Dhondt, I.; Nelis, H.J.; Coenye, T. Involvement of Toxin-Antitoxin Modules in Burkholderia Cenocepacia Biofilm Persistence. Pathog. Dis. 2014, 71, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Berne, C.; Zappa, S.; Brun, Y. V EDNA-Stimulated Cell Dispersion from Caulobacter Crescentus Biofilms upon Oxygen Limitation Is Dependent on a Toxin–Antitoxin System. eLife 2023, 12, e80808. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, K.P.; Chanin, R.B.; Sistrunk, J.R.; Rasko, D.A.; Fink, P.J.; Barry, E.M.; Nataro, J.P.; Faherty, C.S. Analysis of Shigella Flexneri Resistance, Biofilm Formation, and Transcriptional Profile in Response to Bile Salts. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef]
- Wood, T.L.; Wood, T.K. The HigB/HigA Toxin/Antitoxin System of Pseudomonas Aeruginosa Influences the Virulence Factors Pyochelin, Pyocyanin, and Biofilm Formation. Microbiologyopen 2016, 5, 499–511. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, B.; Li, M.; Shi, J.; Long, Y.; Jin, Y.; Bai, F.; Cheng, Z.; Jin, S.; Wu, W. HigB Reciprocally Controls Biofilm Formation and the Expression of Type III Secretion System Genes through Influencing the Intracellular C-Di-GMP Level in Pseudomonas Aeruginosa. Toxins 2018, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhao, Q.; Huang, H.; Fang, Z.; Hu, Y. Edwardsiella Piscicida HigB: A Type II Toxin That Is Essential to Oxidative Resistance, Biofilm Formation, Serum Survival, Intracellular Propagation, and Host Infection. Aquaculture 2021, 535, 736382. [Google Scholar] [CrossRef]
- Hansen, S.; Vulić, M.; Min, J.; Yen, T.-J.; Schumacher, M.A.; Brennan, R.G.; Lewis, K. Regulation of the Escherichia coli HipBA Toxin-Antitoxin System by Proteolysis. PLoS ONE 2012, 7, e39185. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, Q.; Li, M.; Heijstra, B.D.; Wang, S.; Liang, Q.; Qi, Q. Escherichia coli Toxin Gene HipA Affects Biofilm Formation and DNA Release. Microbiology 2013, 159, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xia, K.; Li, P.; Qian, C.; Li, Y.; Liang, X. Functional Investigation of the Chromosomal CcdAB and HipAB Operon in Escherichia coli Nissle 1917. Appl. Microbiol. Biotechnol. 2020, 104, 6731–6747. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Hill, C. Gut Bacteriophage: Current Understanding and Challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- LeRoux, M.; Laub, M.T. Toxin-Antitoxin Systems as Phage Defense Elements. Annu. Rev. Microbiol. 2022, 76, 21–43. [Google Scholar] [CrossRef]
- Hazan, R.; Engelberg-Kulka, H. Escherichia coli MazEF-Mediated Cell Death as a Defense Mechanism That Inhibits the Spread of Phage P1. Mol. Genet. Genom. 2004, 272, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Guegler, C.K.; Laub, M.T. Shutoff of Host Transcription Triggers a Toxin-Antitoxin System to Cleave Phage RNA and Abort Infection. Mol. Cell 2021, 81, 2361–2373.e9. [Google Scholar] [CrossRef]
- Pecota, D.C.; Wood, T.K. Exclusion of T4 Phage by the Hok/Sok Killer Locus from Plasmid R1. J. Bacteriol. 1996, 178, 2044–2050. [Google Scholar] [CrossRef]
- Rakitina, D.V.; Manolov, A.I.; Kanygina, A.V.; Garushyants, S.K.; Baikova, J.P.; Alexeev, D.G.; Ladygina, V.G.; Kostryukova, E.S.; Larin, A.K.; Semashko, T.A.; et al. Genome Analysis of E. coli Isolated from Crohn’s Disease Patients. BMC Genom. 2017, 18, 544. [Google Scholar] [CrossRef] [PubMed]
- Misson, P.; Bruder, E.; Cornuault, J.K.; De Paepe, M.; Nicolas, P.; Demarre, G.; Lakisic, G.; Petit, M.-A.; Espeli, O.; Lecointe, F. Phage Production Is Blocked in the Adherent-Invasive Escherichia coli LF82 upon Macrophage Infection. PLoS Pathog. 2023, 19, e1011127. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, B.; Domingo-Calap, P. Phage Therapy in Gastrointestinal Diseases. Microorganisms 2020, 8, 1420. [Google Scholar] [CrossRef] [PubMed]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages Targeting Adherent Invasive Escherichia coli Strains as a Promising New Treatment for Crohn’s Disease. J. Crohns Colitis 2017, 11, jjw224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, W.; Yao, J.; Wang, X.; Liu, D.; Wang, P. The HipAB Toxin-Antitoxin System Stabilizes a Composite Genomic Island in Shewanella Putrefaciens CN-32. Front. Microbiol. 2022, 13, 858857. [Google Scholar] [CrossRef]
- Yao, X.; Chen, T.; Shen, X.; Zhao, Y.; Wang, M.; Rao, X.; Yin, S.; Wang, J.; Gong, Y.; Lu, S.; et al. The Chromosomal SezAT Toxin-Antitoxin System Promotes the Maintenance of the SsPI-1 Pathogenicity Island in Epidemic Streptococcus Suis. Mol. Microbiol. 2015, 98, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Huguet, K.T.; Gonnet, M.; Doublet, B.; Cloeckaert, A. A Toxin Antitoxin System Promotes the Maintenance of the IncA/C-Mobilizable Salmonella Genomic Island 1. Sci. Rep. 2016, 6, 32285. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.A.F.; Waldor, M.K. A Toxin-Antitoxin System Promotes the Maintenance of an Integrative Conjugative Element. PLoS Genet. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- McVicker, G.; Tang, C.M. Deletion of Toxin–Antitoxin Systems in the Evolution of Shigella Sonnei as a Host-Adapted Pathogen. Nat. Microbiol. 2016, 2, 16204. [Google Scholar] [CrossRef]
- Mathers, A.J.; Peirano, G.; Pitout, J.D.D. The Role of Epidemic Resistance Plasmids and International High-Risk Clones in the Spread of Multidrug-Resistant Enterobacteriaceae. Clin. Microbiol. Rev. 2015, 28, 565–591. [Google Scholar] [CrossRef]
- Pilla, G.; Tang, C.M. Going around in Circles: Virulence Plasmids in Enteric Pathogens. Nat. Rev. Microbiol. 2018, 16, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Chukwudi, C.U.; Good, L. The Role of the Hok/Sok Locus in Bacterial Response to Stressful Growth Conditions. Microb. Pathog. 2015, 79, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Duprilot, M.; Decre, D.; Genel, N.; Drieux, L.; Sougakoff, W.; Arlet, G. Diversity and Functionality of Plasmid-Borne VagCD Toxin-Antitoxin Systems of Klebsiella Pneumoniae. J. Antimicrob. Chemother. 2017, 72, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, P.; Iredell, J.R. Carriage of Type II Toxin-Antitoxin Systems by the Growing Group of IncX Plasmids. Plasmid 2017, 91, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Arcus, V.L.; Mckenzie, J.L.; Robson, J.; Cook, G.M. The PIN-Domain Ribonucleases and the Prokaryotic VapBC Toxin-Antitoxin Array. Protein. Eng. Des. Sel. 2011, 24, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Brendler, T.; Davis, M.; Reaves, L.; Austin, S. Surprising Dependence on Postsegregational Killing of Host Cells for Maintenance of the Large Virulence Plasmid of Shigella Flexneri. J. Bacteriol. 2005, 187, 2768–2773. [Google Scholar] [CrossRef] [PubMed]
- Wurpel, D.J.; Beatson, S.A.; Totsika, M.; Petty, N.K.; Schembri, M.A. Chaperone-Usher Fimbriae of Escherichia coli. PLoS ONE 2013, 8, e52835. [Google Scholar] [CrossRef] [PubMed]
- Ulett, G.C.; Mabbett, A.N.; Fung, K.C.; Webb, R.I.; Schembri, M.A. The Role of F9 Fimbriae of Uropathogenic Escherichia coli in Biofilm Formation. Microbiology 2007, 153, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- Conover, M.S.; Ruer, S.; Taganna, J.; Kalas, V.; De Greve, H.; Pinkner, J.S.; Dodson, K.W.; Remaut, H.; Hultgren, S.J. Inflammation-Induced Adhesin-Receptor Interaction Provides a Fitness Advantage to Uropathogenic E. coli during Chronic Infection. Cell Host Microbe 2016, 20, 482–492. [Google Scholar] [CrossRef]
- Tsuchimoto, S.; Ohtsubo, H.; Ohtsubo, E. Two Genes, PemK and PemI, Responsible for Stable Maintenance of Resistance Plasmid R100. J. Bacteriol. 1988, 170, 1461–1466. [Google Scholar] [CrossRef]
- Bukowski, M.; Lyzen, R.; Helbin, W.M.; Bonar, E.; Szalewska-Palasz, A.; Wegrzyn, G.; Dubin, G.; Dubin, A.; Wladyka, B. A Regulatory Role for Staphylococcus Aureus Toxin–Antitoxin System PemIKSa. Nat. Commun. 2013, 4, 2012. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, I.; Blasco, L.; Pacios, O.; Fernández-García, L.; Ambroa, A.; López, M.; Ortiz-Cartagena, C.; Cuenca, F.F.; Oteo-Iglesias, J.; Pascual, Á.; et al. The Role of PemIK (PemK/PemI) Type II TA System from Klebsiella Pneumoniae Clinical Strains in Lytic Phage Infection. Sci. Rep. 2022, 12, 4488. [Google Scholar] [CrossRef]
- Masuda, Y.; Miyakawa, K.; Nishimura, Y.; Ohtsubo, E. ChpA and ChpB, Escherichia coli Chromosomal Homologs of the Pem Locus Responsible for Stable Maintenance of Plasmid R100. J. Bacteriol. 1993, 175, 6850–6856. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, M.; Hyz, K.; Janczak, M.; Hydzik, M.; Dubin, G.; Wladyka, B. Identification of Novel MazEF/PemIK Family Toxin-Antitoxin Loci and Their Distribution in the Staphylococcus Genus. Sci. Rep. 2017, 7, 13462. [Google Scholar] [CrossRef] [PubMed]
- Janczak, M.; Hyz, K.; Bukowski, M.; Lyzen, R.; Hydzik, M.; Wegrzyn, G.; Szalewska-Palasz, A.; Grudnik, P.; Dubin, G.; Wladyka, B. Chromosomal Localization of PemIK Toxin-Antitoxin System Results in the Loss of Toxicity—Characterization of PemIK-Sp from Staphylococcus Pseudintermedius. Microbiol. Res. 2020, 240, 126529. [Google Scholar] [CrossRef] [PubMed]
- Audoly, G.; Vincentelli, R.; Edouard, S.; Georgiades, K.; Mediannikov, O.; Gimenez, G.; Socolovschi, C.; Mège, J.-L.; Cambillau, C.; Raoult, D. Effect of Rickettsial Toxin VapC on Its Eukaryotic Host. PLoS ONE 2011, 6, e26528. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Rossinyol, Q.; Camprubí-Font, C.; López-Siles, M.; Martinez-Medina, M. Identification of Differences in Gene Expression Implicated in the Adherent-Invasive Escherichia coli Phenotype during in Vitro Infection of Intestinal Epithelial Cells. Front. Cell. Infect. Microbiol. 2023, 13, 1228159. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Patel, P.; Verma, S.K.; Mishra, P.; Sahu, B.R.; Panda, P.K.; Kushwaha, G.S.; Senapati, S.; Misra, N.; Suar, M. The Hha–TomB Toxin–Antitoxin Module in Salmonella Enterica Serovar Typhimurium Limits Its Intracellular Survival Profile and Regulates Host Immune Response. Cell Biol. Toxicol. 2022, 38, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tang, H.; Bao, R. Comparative Analysis of Five Type II TA Systems Identified in Pseudomonas Aeruginosa Reveals Their Contributions to Persistence and Intracellular Survival. Front. Cell. Infect. Microbiol. 2023, 13, 1127786. [Google Scholar] [CrossRef]
- Ren, D.; Walker, A.N.; Daines, D.A. Toxin-Antitoxin Loci VapBC-1 and VapXD Contribute to Survival and Virulence in Nontypeable Haemophilus Influenzae. BMC Microbiol. 2012, 12, 263. [Google Scholar] [CrossRef]
- Ren, D.; Kordis, A.A.; Sonenshine, D.E.; Daines, D.A. The ToxAvapA Toxin-Antitoxin Locus Contributes to the Survival of Nontypeable Haemophilus Influenzae during Infection. PLoS ONE 2014, 9, e91523. [Google Scholar] [CrossRef] [PubMed]
- Hopper, S.; Wilbur, J.S.; Vasquez, B.L.; Larson, J.; Clary, S.; Mehr, I.J.; Seifert, H.S.; So, M. Isolation of Neisseria Gonorrhoeae Mutants That Show Enhanced Trafficking across Polarized T84 Epithelial Monolayers. Infect. Immun. 2000, 68, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of Salmonella by Macrophages Induces Formation of Nonreplicating Persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Cheverton, A.M.; Gollan, B.; Przydacz, M.; Wong, C.T.; Mylona, A.; Hare, S.A.; Helaine, S. A Salmonella Toxin Promotes Persister Formation through Acetylation of TRNA. Mol. Cell 2016, 63, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Rycroft, J.A.; Gollan, B.; Grabe, G.J.; Hall, A.; Cheverton, A.M.; Larrouy-Maumus, G.; Hare, S.A.; Helaine, S. Activity of Acetyltransferase Toxins Involved in Salmonella Persister Formation during Macrophage Infection. Nat. Commun. 2018, 9, 1993. [Google Scholar] [CrossRef] [PubMed]
- Stårsta, M.; Hammarlöf, D.L.; Wäneskog, M.; Schlegel, S.; Xu, F.; Heden Gynnå, A.; Borg, M.; Herschend, S.; Koskiniemi, S. RHS-Elements Function as Type II Toxin-Antitoxin Modules That Regulate Intra-Macrophage Replication of Salmonella Typhimurium. PLoS Genet. 2020, 16, e1008607. [Google Scholar] [CrossRef] [PubMed]
- Michaux, C.; Hartke, A.; Martini, C.; Reiss, S.; Albrecht, D.; Budin-Verneuil, A.; Sanguinetti, M.; Engelmann, S.; Hain, T.; Verneuil, N.; et al. Involvement of Enterococcus Faecalis Small RNAs in Stress Response and Virulence. Infect. Immun. 2014, 82, 3599–3611. [Google Scholar] [CrossRef] [PubMed]
- Sonika, S.; Singh, S.; Mishra, S.; Verma, S. Toxin-Antitoxin Systems in Bacterial Pathogenesis. Heliyon 2023, 9, e14220. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent Bacterial Infections and Persister Cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Bruder, E.; Espéli, O. Escherichia coli Bacteria Associated with Crohn’s Disease Persist within Phagolysosomes. Curr. Opin. Microbiol. 2022, 70, 102206. [Google Scholar] [CrossRef]
- Korch, S.B.; Henderson, T.A.; Hill, T.M. Characterization of the HipA7 Allele of Escherichia coli and Evidence That High Persistence Is Governed by (p)PpGpp Synthesis. Mol. Microbiol. 2003, 50, 1199–1213. [Google Scholar] [CrossRef]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active Starvation Responses Mediate Antibiotic Tolerance in Biofilms and Nutrient-Limited Bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Podlesek, Z.; Žgur Bertok, D. The DNA Damage Inducible SOS Response Is a Key Player in the Generation of Bacterial Persister Cells and Population Wide Tolerance. Front. Microbiol. 2020, 11, 561210. [Google Scholar] [CrossRef]
- Kim, J.S.; Wood, T.K. Persistent Persister Misperceptions. Front. Microbiol. 2016, 7, 2134. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of Bacterial Persistence during Stress and Antibiotic Exposure. Science 2016, 354, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Fino, C.; Sørensen, M.A.; Semsey, S.; Gerdes, K. Prophages and Growth Dynamics Confound Experimental Results with Antibiotic-Tolerant Persister Cells. mBio 2017, 8, e01964-17. [Google Scholar] [CrossRef] [PubMed]
- Moyed, H.S.; Bertrand, K.P. HipA, a Newly Recognized Gene of Escherichia coli K-12 That Affects Frequency of Persistence after Inhibition of Murein Synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [CrossRef]
- Kaspy, I.; Rotem, E.; Weiss, N.; Ronin, I.; Balaban, N.Q.; Glaser, G. HipA-Mediated Antibiotic Persistence via Phosphorylation of the Glutamyl-TRNA-Synthetase. Nat. Commun. 2013, 4, 3001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Eckart, M.R.; Shapiro, L. A Bacterial Toxin Perturbs Intracellular Amino Acid Balance To Induce Persistence. mBio 2021, 12, e03020-20. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Wade, W.D.; Akierman, S.; Vacchi-Suzzi, C.; Stremick, C.A.; Turner, R.J.; Ceri, H. The Chromosomal Toxin Gene YafQ Is a Determinant of Multidrug Tolerance for Escherichia coli Growing in a Biofilm. Antimicrob. Agents Chemother. 2009, 53, 2253–2258. [Google Scholar] [CrossRef]
- Dörr, T.; Vulić, M.; Lewis, K. Ciprofloxacin Causes Persister Formation by Inducing the TisB Toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef]
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; Van den Bergh, B.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and Membrane Depolarization Are Part of a Microbial Bet-Hedging Strategy That Leads to Antibiotic Tolerance. Mol. Cell 2015, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wood, T.K. Toxins Hha and CspD and Small RNA Regulator Hfq Are Involved in Persister Cell Formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 2010, 391, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Dufour, D.; Mankovskaia, A.; Chan, Y.; Motavaze, K.; Gong, S.; Lévesque, C.M. A Tripartite Toxin-antitoxin Module Induced by Quorum Sensing Is Associated with the Persistence Phenotype in Streptococcus Mutans. Mol. Oral. Microbiol. 2018, 33, 420–429. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.; Ghodasra, M.; Schardt, J.; Quan, D.; Pottash, A.E.; Shang, W.; Jay, S.M.; Payne, G.F.; Chang, M.W.; March, J.C.; et al. A Platform of Genetically Engineered Bacteria as Vehicles for Localized Delivery of Therapeutics: Toward Applications for Crohn’s Disease. Bioeng. Transl. Med. 2018, 3, 209–221. [Google Scholar] [CrossRef]
- LeRoux, M.; Culviner, P.H.; Liu, Y.J.; Littlehale, M.L.; Laub, M.T. Stress Can Induce Transcription of Toxin-Antitoxin Systems without Activating Toxin. Mol. Cell 2020, 79, 280–292.e8. [Google Scholar] [CrossRef]

{kind=link}
| TA ID | Toxin | Antitoxin | Family/Domain | Comments |
|---|---|---|---|---|
| TA214828 | K8B90_RS03460 (symE) | -(symR) | symER/SymE (toxin) | Type I |
| TA214832 | K8B90_RS03850 (hokC) | -(sokC) | hok-sok/- | Type I |
| TA214852 | K8B90_RS22020 (ldrD) | -(rdlD) | ldrD-rdlD/Ldr (toxin) | Type I |
| TA214826 | K8B90_RS00850 (higB) | K8B90_RS00855 (higA) | higBA (relBE)/HTH (antitoxin) | Type II |
| TA214834 | K8B90_RS04020 (ccdB) | K8B90_RS04015 (ccdA) | ccdAB/CcdA (antitoxin) | Type II |
| TA214835 | K8B90_RS05120 (yafO) | K8B90_RS05115 (yafN) | yafN-yafO (relBE)/YafO-YafN | Type II |
| TA214836 | K8B90_RS06040 (Hha) | K8B90_RS06045 (TomB) | Hha-TomB/- | Type II |
| TA214837 | K8B90_RS11255 (higB) | K8B90_RS11250 (higA) | higBA (relBE)/HigB-HigA | Type II |
| TA214838 | K8B90_RS11415 (hipA) | K8B90_RS11420 (hipB) | hipBA/HipA-HipB | Type II; related to MGE |
| TA214847 | K8B90_RS14005 (pemK) | K8B90_RS14010 (pemI) | pemIK/PRK09812-MazE | Type II; related to MGE |
| TA214848 | K8B90_RS14290 (yoeB) | K8B90_RS14295 (yefM) | yefM-yoeB (relBE)/YoeB-YefM | Type II |
| TA214849 | K8B90_RS17940 (mazF) | K8B90_RS17945 (mazE) | mazEF/PRK09907-MazE | Type II |
| TA214851 | K8B90_RS19925 (yhaV) | K8B90_RS19920 (prlF) | prlF-yhaV (relBE)/YhaV-PrlF | Type II |
| TA214850 | K8B90_RS18545 (cptA) | K8B90_RS18550 (cptB) | cptAB/CptA (toxin) | Type IV |
| TA214827 | K8B90_RS02180 (ghoT) | K8B90_RS02175 (ghoS) | ghoTS/ghoT-GhoS | Type V |
| TA214839 | -(SdsR) | -(RyeA) | SdsR-RyeA/- | Type VIII |
| TA ID | Toxin | Antitoxin | Family/Domain | Comments |
|---|---|---|---|---|
| TA027329 | NRG857_RS00075 (hokC) | -(sokC) | hok-sok/- | Type I; TA1 at [9] |
| TA027349 | NRG857_RS17965 (ldrD) | -(rdlD) | ldrD-rdlD/Ldr (toxin) | Type I; TA14 at [9] |
| TA027353 | NRG857_RS22450 (symE) | -(symR) | symER/SymE (toxin) | Type I; TA16 at [9] |
| TA027597 | NRG857_RS23200 (srnB) | -(sok) | hok-sok/- | Type I; on plasmid pO83_CORR |
| TA027602 | NRG857_RS23320 (hok) | -(sok) | hok-sok/- | Type I; on plasmid pO83_CORR |
| TA027331 | NRG857_RS00245 (ccdB) | NRG857_RS00240 (ccdA) | ccdAB/CcdA (antitoxin) | Type II; TA17 at [9] |
| TA027332 | NRG857_RS01300 (yafO) | NRG857_RS01295 (yafN) | yafN-yafO (relBE)/YafO-YafN | Type II; TA18 at [9] |
| TA027333 | NRG857_RS02210 (Hha) | NRG857_RS02215 (TomB) | Hha-TomB/- | Type II |
| TA027334 | NRG857_RS07485 (higB) | NRG857_RS07480 (higA) | higBA (relBE)/HigB-HigA | Type II; TA20 at [9] |
| TA027335 | NRG857_RS07640 (hipA) | NRG857_RS07645 (hipB) | hipBA/HipA-HipB | Type II; TA21 at [9]; related to MGE |
| TA027344 | NRG857_RS10225 (pemK) | NRG857_RS10230 (pemI) | pemIK/PRK09812-MazE | Type II; TA22 at [9]; related to MGE |
| TA027345 | NRG857_RS10510 (yoeB) | NRG857_RS10515 (yefM) | yefM-yoeB (relBE)/YoeB-YefM | Type II; TA23 at [9] |
| TA027346 | NRG857_RS13925 (mazF) | NRG857_RS13930 (mazE) | mazEF/PRK09907-MazE | Type II; TA24 at [9] |
| TA027348 | NRG857_RS15890 (yhaV) | NRG857_RS15885 (prlF) | prlF-yhaV (relBE)/YhaV-PrlF | Type II; TA25 at [9] |
| TA027351 | NRG857_RS19860 (higB) | NRG857_RS19865 (higA) | higBA (relBE)/HTH (antitoxin) | Type II; TA27 at [9] |
| TA027596 | NRG857_RS22890 (vagD) | NRG857_RS22885 (vagC) | vagCD/VapC-VagC | Type II; on the plasmid pO83_CORR |
| TA027601 | NRG857_RS23235 (vapC) | NRG857_RS23230 (vapB) | vapBC/VapC-VagC | Type II; on the plasmid pO83_CORR |
| TA027347 | NRG857_RS14535 (cptA) | NRG857_RS14540 (cptB) | cptAB/CptA (toxin) | Type IV; TA31 at [9] |
| TA027352 | NRG857_RS21190 (ghoT) | NRG857_RS21185 (ghoS) | ghoTS/ghoT-GhoS | Type V; TA32 at [9] |
| TA027336 | 1894846..1894948 (-) -(SdsR) | -(RyeA) | SdsR-RyeA/- | Type VIII |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante, P.; Ramos-Corominas, M.N.; Martinez-Medina, M. Contribution of Toxin–Antitoxin Systems to Adherent-Invasive E. coli Pathogenesis. Microorganisms 2024, 12, 1158. https://doi.org/10.3390/microorganisms12061158
Bustamante P, Ramos-Corominas MN, Martinez-Medina M. Contribution of Toxin–Antitoxin Systems to Adherent-Invasive E. coli Pathogenesis. Microorganisms. 2024; 12(6):1158. https://doi.org/10.3390/microorganisms12061158
Chicago/Turabian StyleBustamante, Paula, María Núria Ramos-Corominas, and Margarita Martinez-Medina. 2024. "Contribution of Toxin–Antitoxin Systems to Adherent-Invasive E. coli Pathogenesis" Microorganisms 12, no. 6: 1158. https://doi.org/10.3390/microorganisms12061158
APA StyleBustamante, P., Ramos-Corominas, M. N., & Martinez-Medina, M. (2024). Contribution of Toxin–Antitoxin Systems to Adherent-Invasive E. coli Pathogenesis. Microorganisms, 12(6), 1158. https://doi.org/10.3390/microorganisms12061158