
Thanos: An R Package for the Gene-Centric Analysis of Functional Potential in Metagenomic Samples

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Importing Depths Files
2.2. Running HMMER
2.3. Aggregation and Normalization
2.4. Visualization
2.5. Parallelization
2.6. Dependencies
3. Results
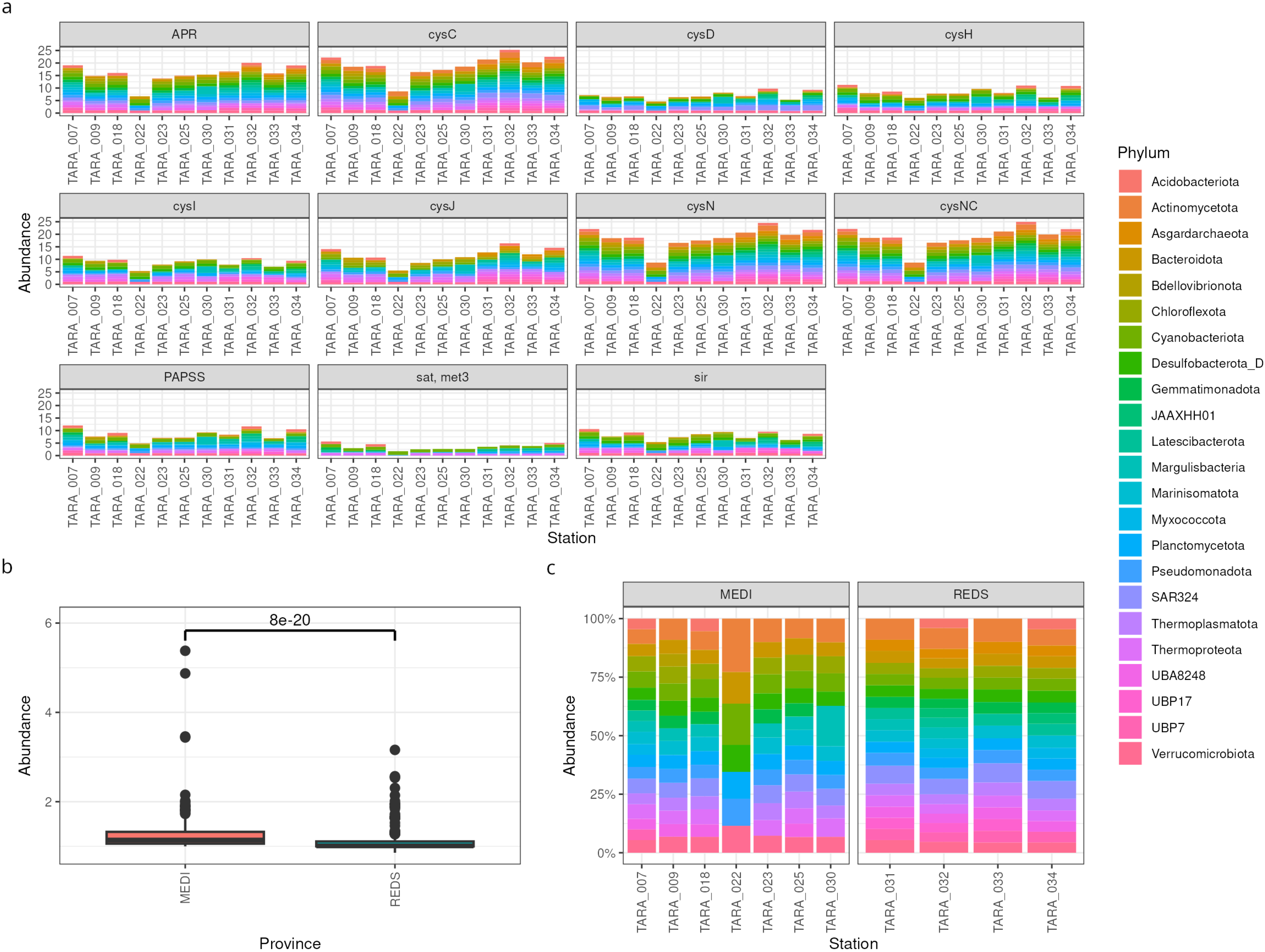
3.1. MAGs Workflow: Sulfur Metabolism by Taxonomy
3.2. Contigs Workflow: Prevalence of Glycolysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| MAG | Metagenome-Assembled Genome |
| OTU | Operational Taxonomic Unit |
| HMM | Hidden Markov Model |
| KEGG | Kyoto Encyclopedia of Gene Elements |
References
- Zhang, L.; Chen, F.; Zeng, Z.; Xu, M.; Sun, F.; Yang, L.; Bi, X.; Lin, Y.; Gao, Y.; Hao, H.; et al. Advances in metagenomics and its application in environmental microorganisms. Front. Microbiol. 2021, 12, 766364. [Google Scholar] [CrossRef]
- Afzaal, M.; Saeed, F.; Shah, Y.A.; Hussain, M.; Rabail, R.; Socol, C.T.; Hassoun, A.; Pateiro, M.; Lorenzo, J.M.; Rusu, A.V.; et al. Human gut microbiota in health and disease: Unveiling the relationship. Front. Microbiol. 2022, 13, 999001. [Google Scholar] [CrossRef]
- Borroni, D.; Paytuví-Gallart, A.; Sanseverino, W.; Gómez-Huertas, C.; Bonci, P.; Romano, V.; Giannaccare, G.; Rechichi, M.; Meduri, A.; Oliverio, G.W.; et al. Exploring the healthy eye microbiota niche in a multicenter study. Int. J. Mol. Sci. 2022, 23, 10229. [Google Scholar] [CrossRef]
- Rocha-de Lossada, C.; Mazzotta, C.; Gabrielli, F.; Papa, F.T.; Gómez-Huertas, C.; García-López, C.; Urbinati, F.; Rachwani-Anil, R.; García-Lorente, M.; Sánchez-González, J.M.; et al. Ocular surface microbiota in naïve keratoconus: A multicenter validation study. J. Clin. Med. 2023, 12, 6354. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Coordinators, T.O.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E.; et al. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput. Struct. Biotechnol. J. 2021, 19, 6301–6314. [Google Scholar] [CrossRef]
- Konopka, A. What is microbial community ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.L.; Xu, L.; Cui, L.; Fu, G.Y.; Xu, X.W. Metagenome-based analysis of carbon-fixing microorganisms and their carbon-fixing pathways in deep-sea sediments of the southwestern Indian Ocean. Mar. Genom. 2023, 70, 101045. [Google Scholar] [CrossRef]
- Milanese, A.; Mende, D.R.; Paoli, L.; Salazar, G.; Ruscheweyh, H.J.; Cuenca, M.; Hingamp, P.; Alves, R.; Costea, P.I.; Coelho, L.P.; et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Prokka: Rapid Prokaryotic Genome Annotation. Available online: https://github.com/tseemann/prokka (accessed on 17 June 2024).
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- EggNOG-Mapper: Genome-Wide Functional Annotation. Available online: http://eggnog-mapper.embl.de/ (accessed on 17 June 2024).
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 17 June 2024).
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Use R!), 2nd ed.; Springer: Cham, Switzerland, 2016; 276p. [Google Scholar]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- MetaBAT: A Robust Statistical Framework for Reconstructing Genomes from Metagenomic Data. Available online: https://bitbucket.org/berkeleylab/metabat/src/master/ (accessed on 17 June 2024).
- wwood/CoverM: Read Coverage Calculator for Metagenomics. Available online: https://github.com/wwood/CoverM (accessed on 17 June 2024).
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Prodigal Gene Prediction Software. Available online: https://github.com/hyattpd/Prodigal (accessed on 17 June 2024).
- Krakau, S.; Straub, D.; Gourlé, H.; Gabernet, G.; Nahnsen, S. nf-core/mag: A best-practice pipeline for metagenome hybrid assembly and binning. NAR Genom. Bioinform. 2022, 4, lqac007. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- nf-core/mag: Assembly and Binning of Metagenomes. Available online: https://nf-co.re/mag/2.5.4 (accessed on 17 June 2024).
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.M. A grpE mutant of Escherichia coli is more resistant to heat than the wild-type. J. Gen. Microbiol. 1990, 136, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 17 June 2024).
- Sunagawa, S.; Coelho, L.; Chaffron, S.; Kultima, J.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tárraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The european nucleotide archive. Nucleic Acids Res. 2011, 39, D28–D31. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, R.F.; Deeley, R.G.; Mullinix, K.P. Regulation of gene expression in prokaryotic organisms. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 1976; Volume 18, pp. 1–67. [Google Scholar] [CrossRef]
- Allen, K.J.; Lepp, D.; McKellar, R.C.; Griffiths, M.W. Examination of stress and virulence gene expression in Escherichia coli O157:H7 using targeted microarray analysis. Foodborne Pathog. Dis. 2008, 5, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Echtenkamp, P.L.; Wilson, D.B.; Shuler, M.L. Cell cycle progression in Escherichia coli B/r affects transcription of certain genes: Implications for synthetic genome design. Biotechnol. Bioeng. 2009, 102, 902–909. [Google Scholar] [CrossRef]
- Côté, J.P.; French, S.; Gehrke, S.S.; MacNair, C.R.; Mangat, C.S.; Bharat, A.; Brown, E.D. The Genome-Wide Interaction Network of Nutrient Stress Genes in Escherichia coli. mBio 2016, 7, e01714-16. [Google Scholar] [CrossRef]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Brockhurst, M.A.; Harrison, E.; Hall, J.P.J.; Richards, T.; McNally, A.; MacLean, C. The ecology and evolution of pangenomes. Curr. Biol. 2019, 29, R1094–R1103. [Google Scholar] [CrossRef] [PubMed]
- Gally, D.L.; Stevens, M.P. Microbe Profile: Escherichia coli O157:H7—Notorious relative of the microbiologist’s workhorse. Microbiology 2017, 163, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zou, S.; Zhang, H.; Liu, Q.; Song, Z.; Huang, Y.; Hu, X. Genome-resolved metagenomics provides insights into the microbial-mediated sulfur and nitrogen cycling in temperate seagrass meadows. Front. Mar. Sci. 2023, 10, 1245288. [Google Scholar] [CrossRef]
- Wang, J.; Jia, H. Metagenome-wide association studies: Fine-mining the microbiome. Nat. Rev. Microbiol. 2016, 14, 508–522. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Marotta, F.; Wu, M. Thanos: An R Package for the Gene-Centric Analysis of Functional Potential in Metagenomic Samples. Microorganisms 2024, 12, 1264. https://doi.org/10.3390/microorganisms12071264
Zhao Z, Marotta F, Wu M. Thanos: An R Package for the Gene-Centric Analysis of Functional Potential in Metagenomic Samples. Microorganisms. 2024; 12(7):1264. https://doi.org/10.3390/microorganisms12071264
Chicago/Turabian StyleZhao, Zhe, Federico Marotta, and Min Wu. 2024. "Thanos: An R Package for the Gene-Centric Analysis of Functional Potential in Metagenomic Samples" Microorganisms 12, no. 7: 1264. https://doi.org/10.3390/microorganisms12071264
APA StyleZhao, Z., Marotta, F., & Wu, M. (2024). Thanos: An R Package for the Gene-Centric Analysis of Functional Potential in Metagenomic Samples. Microorganisms, 12(7), 1264. https://doi.org/10.3390/microorganisms12071264